Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements


 ,
,  ,
, 



Abstract
1. Introduction
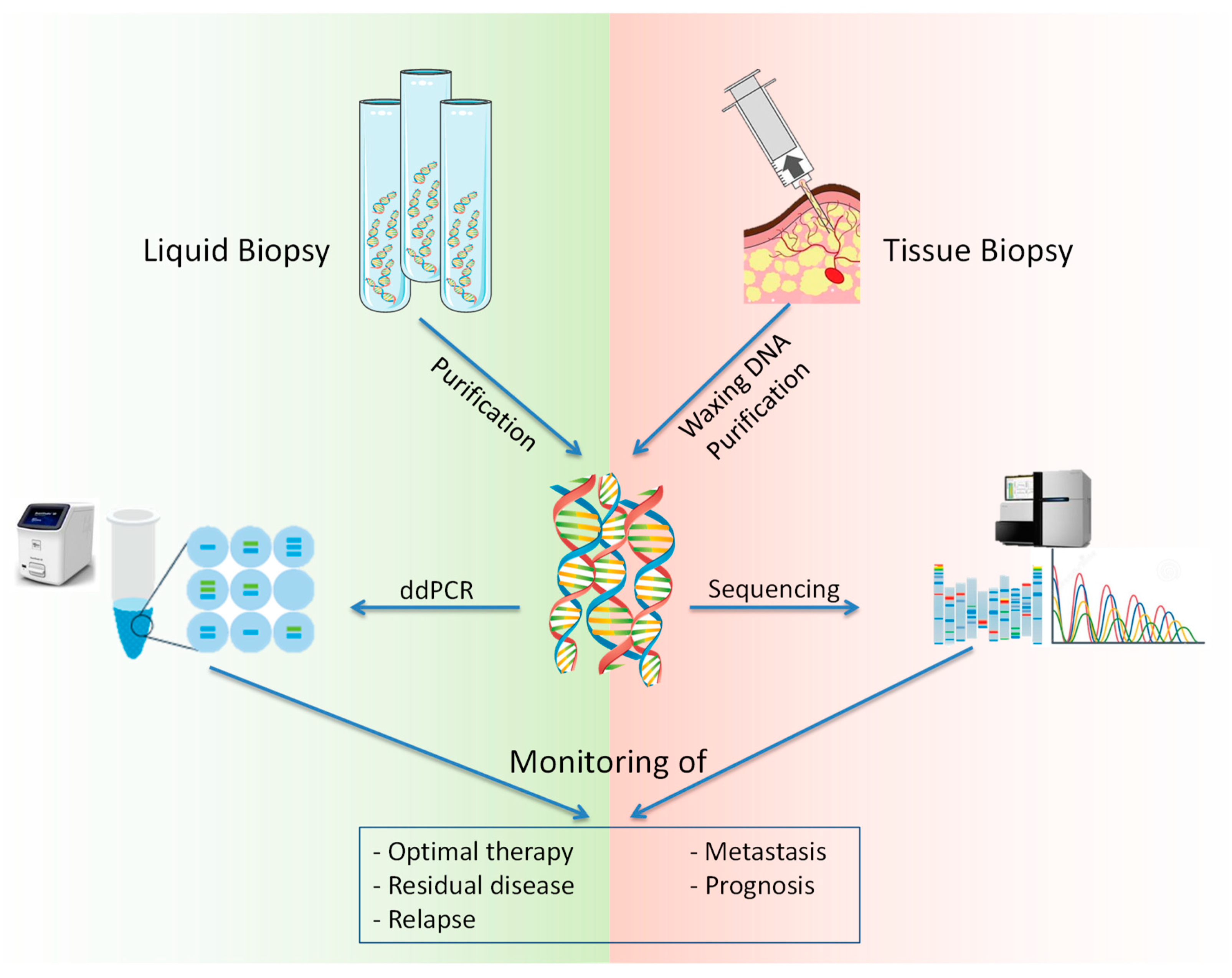
2. NGS, ddPCR, and Liquid Biopsy for ctDNA Analysis
3. Epigenetic Modifications of Circulating DNA May Reflect Tissue Origin of an Unknown Primary Cancer
4. Circulating DNA as a Prognostic Criterion in Oncology
5. Circulating Tumor DNA Analysis for Cancer Personalized Medicine
6. Circulating DNA in Glioma Diagnosis
7. Urinary Circulating DNA in Urological Tumors
8. Multiplex Genotyping Based on ctDNA Mass-Spectrometry
9. Limitations of the Use of ctDNA in Cancer Routine Practice
- The majority of patients in the study had stage II–III of disease, which does not yet provide an opportunity to assess the applicability of the test for early diagnosis [232].
- As control, the authors analyzed the urine of only healthy individuals. Thus, the high specificity of the CancerSeek approach requires further validation with non-cancer control with associated diseases, such as inflammatory diseases of the genitourinary system, which are common in the elderly [233].
- The limitations of the method are linked with the same heterogeneity of tumors, such as lung cancer [234]. These circumstances cannot be attributed to the shortcomings of the method, but make it necessary to add additional markers that may increase the accuracy of the study, such as RNA of exosomes for early diagnosis of non-small cell lung cancer [235].
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Aucamp, J.; Bronkhorst, A.J.; Badenhorst, C.P.; Pretorius, P.J. A historical and evolutionary perspective on the biological significance of circulating DNA and extracellular vesicles. Cell Mol. Life Sci. 2016, 73, 4355–4381. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Nielsen, K.M.; Johnsen, P.J.; Bensasson, D.; Daffonchio, D. Release and persistence of extracellular DNA in the environment. Env. Biosaf. Res. 2007, 6, 37–53. [Google Scholar] [CrossRef]
- Jiang, P.; Lo, Y.M.D. The Long and Short of Circulating Cell-Free DNA and the Ins and Outs of Molecular Diagnostics. Trends Genet. 2016, 32, 360–371. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef]
- Kosovec, J.E.; Zaidi, A.H.; Pounardjian, T.S.; Jobe, B.A. The Potential Clinical Utility of Circulating Tumor DNA in Esophageal Adenocarcinoma: From Early Detection to Therapy. Front. Oncol. 2018, 8, 610. [Google Scholar] [CrossRef]
- Wang, J.; Bettegowda, C. Applications of DNA-based liquid biopsy for central nervous system neoplasms. J. Mol. Diagn. 2017, 19, 24–34. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta 2016, 1863, 157–165. [Google Scholar] [CrossRef]
- Hyun, K.A.; Koo, G.B.; Han, H.; Sohn, J.; Choi, W.; Kim, S.I.; Jung, H.I.; Kim, Y.S. Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget 2016, 7, 24677–24687. [Google Scholar] [CrossRef]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef]
- Togneri, F.S.; Ward, D.G.; Foster, J.M.; Devall, A.J.; Wojtowicz, P.; Alyas, S.; Vasques, F.R.; Oumie, A.; James, N.D.; Cheng, K.K.; et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur. J. Hum. Genet. 2016, 24, 1167–1174. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef]
- Salvi, S.; Casadio, V.; Conteduca, V.; Burgio, S.L.; Menna, C.; Bianchi, E.; Rossi, L.; Carretta, E.; Masini, C.; Amadori, D.; et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br. J. Cancer 2015, 112, 1717–1724. [Google Scholar] [CrossRef]
- Schütz, E.; Akbari, M.R.; Beck, J.; Urnovitz, H.; Zhang, W.W.; Bornemann-Kolatzki, K.; Mitchell, W.M.; Nam, R.K.; Narod, S.A. Chromosomal instability in cell-free DNA is a serum biomarker for prostate cancer. Clin. Chem. 2015, 61, 239–248. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Pantel, K. Circulating DNA as biomarker in breast cancer. Breast Cancer Res. 2015, 17, 136. [Google Scholar] [CrossRef]
- Falzone, L.; Lupo, G.; La Rosa, G.R.M.; Crimi, S.; Anfuso, C.D.; Salemi, R.; Rapisarda, E.; Libra, M.; Candido, S. Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 2019, 11, 610. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Y.; Ye, R.; Zhang, D.; Li, Q.; An, D.; Fang, L.; Lin, Y.; Hou, Y.; Xu, A.; et al. An epigenetic biomarker combination of PCDH17 and POU4F2 detects bladder cancer accurately by methylation analyses of urine sediment DNA in Han Chinese. Oncotarget 2016, 7, 2754–2764. [Google Scholar] [CrossRef]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef]
- Marzese, D.M.; Hirose, H.; Hoon, D.S. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Expert Rev. Mol. Diagn. 2013, 13, 827–844. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Laird, P.W. Early detection: The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Chu, D.; Park, B.H. Liquid biopsy: Unlocking the potentials of cell-free DNA. Virchows Archiv. 2017, 471, 147–154. [Google Scholar] [CrossRef]
- Macías, M.; Alegre, E.; Díaz-Lagares, A.; Patiño, A.; Pérez-Gracia, J.L.; Sanmamed, M.; López-López, R.; Varo, N.; González, A. Liquid Biopsy: From Basic Research to Clinical Practice. Adv. Clin. Chem. 2018, 83, 73–119. [Google Scholar]
- Rothé, F.; Laes, J.F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pellè, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Khier, S.; Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: Critical appraisal of the literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef]
- Di Meo, A.; Bartlett, J.; Cheng, Y.; Pasic, M.D.; Yousef, G.M. Liquid biopsy: A step forward towards precision medicine in urologic malignancies. Mol. Cancer 2017, 16, 80. [Google Scholar] [CrossRef]
- Qin, Z.; Ljubimov, V.A.; Zhou, C.; Tong, Y.; Liang, J. Cell-free circulating tumor DNA in cancer. Chin. J. Cancer 2016, 35, 36. [Google Scholar] [CrossRef]
- Riethdorf, S.; Soave, A.; Rink, M. The current status and clinical value of circulating tumor cells and circulating cell-free tumor DNA in bladder cancer. Transl. Androl. Urol. 2017, 6, 1090. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Woff, E.; Kehagias, P.; Vandeputte, C.; Ameye, L.; Guiot, T.; Paesmans, M.; Hendlisz, A.; Flamen, P. Combining 18F-FDG PET/CT-based Metabolically Active Tumor Volume and Circulating Cell-free DNA Significantly Improves Outcome Prediction in Chemorefractory Metastatic Colorectal Cancer. J. Nucl. Med. 2019. [CrossRef]
- Winther-Larsen, A.; Fledelius, J.; Demuth, C.; Bylov, C.M.; Meldgaard, P.; Sorensen, B.S. Early Change in FDG-PET Signal and Plasma Cell-Free DNA Level Predicts Erlotinib Response in EGFR Wild-Type NSCLC Patients. Transl. Oncol. 2016, 9, 505–511. [Google Scholar] [CrossRef][Green Version]
- Nygaard, A.D.; Holdgaard, P.C.; Spindler, K.L.; Pallisgaard, N.; Jakobsen, A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br. J. Cancer 2014, 110, 363–368. [Google Scholar] [CrossRef]
- Jia, N.; Sun, Z.; Gao, X.; Cheng, Y.; Zhou, Y.; Shen, C.; Chen, W.; Wang, X.; Shi, R.; Li, N.; et al. Serial Monitoring of Circulating Tumor DNA in Patients With Metastatic Colorectal Cancer to Predict the Therapeutic Response. Front. Genet. 2019, 10, 470. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Finotti, A.; Allegretti, M.; Gasparello, J.; Giacomini, P.; Spandidos, D.A.; Spoto, G.; Gambari, R. Liquid biopsy and PCR-free ultrasensitive detection systems in oncology (Review). Int. J. Oncol. 2018, 53, 1395–1434. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Zill, O.A.; Greene, C.; Sebisanovic, D.; Siew, L.M.; Leng, J.; Vu, M.; Hendifar, A.E.; Wang, Z.; Atreya, C.E.; Kelley, R.K.; et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015, 5, 1040–1048. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef]
- Ade, C.; Roy-Engel, A.M.; Deininger, P.L. Alu elements: An intrinsic source of human genome instability. Curr. Opin. Virol. 2013, 3, 639–645. [Google Scholar] [CrossRef]
- Szymañska, K.; Chen, J.G.; Cui, Y.; Gong, Y.Y.; Turner, P.C.; Villar, S.; Wild, C.P.; Parkin, D.M.; Hainaut, P. TP53 R249S mutations, exposure to aflatoxin, and occurrence of hepatocellular carcinoma in a cohort of chronic hepatitis B virus carriers from Qidong, China. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1638–1643. [Google Scholar] [CrossRef]
- Shaw, J.A.; Page, K.; Blighe, K.; Hava, N.; Guttery, D.; Ward, B.; Brown, J.; Ruangpratheep, C.; Stebbing, J.; Payne, R.; et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2012, 22, 220–231. [Google Scholar] [CrossRef]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.H.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Kawakami, H.; Okamoto, I.; Arao, T.; Okamoto, W.; Matsumoto, K.; Taniguchi, H.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K.; et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget 2013, 4, 9–17. [Google Scholar] [CrossRef]
- Shoda, K.; Ichikawa, D.; Fujita, Y.; Masuda, K.; Hiramoto, H.; Hamada, J.; Arita, T.; Konishi, H.; Komatsu, S.; Shiozaki, A.; et al. Monitoring the HER2 copy number status in circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer 2017, 20, 126–135. [Google Scholar] [CrossRef]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef]
- Leary, R.J.; Kinde, I.; Diehl, F.; Schmidt, K.; Clouser, C.; Duncan, C.; Antipova, A.; Lee, C.; McKernan, K.; De La Vega, F.M.; et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci. Transl. Med. 2010, 2, 20ra14. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Gao, X.; Tan, B.H.; Sugrue, R.J.; Tang, K. MALDI mass spectrometry for nucleic acid analysis. In Applications of MALDI-TOF Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2012; pp. 55–77. [Google Scholar]
- Patel, K.M.; van der Vos, K.E.; Smith, C.G.; Mouliere, F.; Tsui, D.; Morris, J.; Chandrananda, D.; Marass, F.; van den Broek, D.; Neal, D.E.; et al. Association Of Plasma And Urinary Mutant DNA With Clinical Outcomes In Muscle Invasive Bladder Cancer. Sci. Rep 2017, 7, 5554. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Wood-Bouwens, C.M.; Ji, H.P. Single Color Multiplexed ddPCR Copy Number Measurements and Single Nucleotide Variant Genotyping. In Digital PCR; Humana Press: New York, NY, USA, 2018; pp. 323–333. [Google Scholar]
- Schiffman, J.D.; Fisher, P.G.; Gibbs, P. Early detection of cancer: Past, present, and future. Am. Soc. Clin. Oncol. Educ. Book 2015, 57–65. [Google Scholar] [CrossRef]
- Gates, T.J. Screening for cancer: Concepts and controversies. Am. Fam. Physician 2014, 90, 625–631. [Google Scholar] [CrossRef]
- Smith, R.A.; Andrews, K.S.; Brooks, D.; Fedewa, S.A.; Manassaram-Baptiste, D.; Saslow, D.; Brawley, O.W.; Wender, R.C. Cancer screening in the United States, 2018: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2018, 68, 297–316. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Bai, Y.; Zhao, H. Liquid biopsy in tumors: Opportunities and challenges. Ann. Transl. Med. 2018, 6, S89. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Anderson, M.W.; Schrijver, I. Next generation DNA sequencing and the future of genomic medicine. Genes 2010, 1, 38–69. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Dawson, S.J. Circulating tumor cells and circulating tumor DNA for precision medicine: Dream or reality? Ann. Oncol. 2014, 25, 2304–2313. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Arakelyan, A.S.; Galeeva, A.K.; Sushhentsev, N.A.; Degtyarevskaya, T.Y.; Chebyshev, N.V. Prospects for the study of circulating in the blood of tumor DNA as a marker of the state of malignant tumors. In Proceedings of the XXII Youth Scientific Forum: Natural and Medical Sciences, Moscow, Russia, 13–17 April 2015. [Google Scholar]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef]
- Mehrotra, M.; Singh, R.R.; Chen, W.; Huang, R.S.P.; Almohammedsalim, A.A.; Barkoh, B.A.; Simien, C.M.; Hernandez, M.; Behrens, C.; Patel, K.P.; et al. Study of Preanalytic and Analytic Variables for Clinical Next-Generation Sequencing of Circulating Cell-Free Nucleic Acid. J. Mol. Diagn. 2017, 19, 514–524. [Google Scholar] [CrossRef]
- Kameta, E.; Sugimori, K.; Kaneko, T.; Ishii, T.; Miwa, H.; Sato, T.; Ishii, Y.; Sue, S.; Sasaki, T.; Yamashita, Y.; et al. Diagnosis of pancreatic lesions collected by endoscopic ultrasound-guided fine-needle aspiration using next-generation sequencing. Oncol. Lett. 2016, 12, 3875–3881. [Google Scholar] [CrossRef]
- Kaiserm, J. ‘Liquid biopsy’ for cancer promises early detection. Science 2018, 359, 259. [Google Scholar] [CrossRef]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef]
- Xu, T.; Kang, X.; You, X.; Dai, L.; Tian, D.; Yan, W.; Yang, Y.; Xiong, H.; Liang, Z.; Zhao, G.Q.; et al. Cross-Platform Comparison of Four Leading Technologies for Detecting EGFR Mutations in Circulating Tumor DNA from Non-Small Cell Lung Carcinoma Patient Plasma. Theranostics 2017, 7, 1437–1446. [Google Scholar] [CrossRef]
- Battaglia, R.; Palini, S.; Vento, M.E.; La Ferlita, A.; Lo Faro, M.J.; Caroppo, E.; Borzì, P.; Falzone, L.; Barbagallo, D.; Ragusa, M.; et al. Identification of extracellular vesicles and characterization of miRNA expression profiles in human blastocoel fluid. Sci. Rep. 2019, 9, 84. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, B.; Tong, X.; Wang, Y.; Wang, C.; Jin, J.; Tian, P.; Li, W. Diagnostic accuracy of droplet digital PCR for detection of EGFR T790M mutation in circulating tumor DNA. Cancer Manag. Res. 2018, 10, 1209–1218. [Google Scholar] [CrossRef]
- Van Ginkel, J.H.; Huibers, M.M.H.; van Es, R.J.J.; de Bree, R.; Willems, S.M. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer 2017, 17, 428. [Google Scholar] [CrossRef]
- Medford, A.J.; Gillani, R.N.; Park, B.H. Detection of Cancer DNA in Early Stage and Metastatic Breast Cancer Patients. Methods Mol. Biol. 2018, 1768, 209–227. [Google Scholar]
- Lund, H.L.; Hughesman, C.B.; McNeil, K.; Clemens, S.; Hocken, K.; Pettersson, R.; Karsan, A.; Foster, L.J.; Haynes, C. Initial diagnosis of chronic myelogenous leukemia based on quantification of M-BCR status using droplet digital PCR. Anal. Bioanal. Chem. 2016, 408, 1079–1094. [Google Scholar] [CrossRef]
- Rapisarda, V.; Ledda, C.; Matera, S.; Fago, L.; Arrabito, G.; Falzone, L.; Marconi, A.; Libra, M.; Loreto, C. Absence of t(14;18) chromosome translocation in agricultural workers after short-term exposure to pesticides. Mol. Med. Rep. 2017, 15, 3379–3382. [Google Scholar] [CrossRef]
- Fenga, C.; Gangemi, S.; Di Salvatore, V.; Falzone, L.; Libra, M. Immunological effects of occupational exposure to lead (Review). Mol. Med. Rep. 2017, 15, 3355–3360. [Google Scholar] [CrossRef]
- Rapisarda, V.; Salemi, R.; Marconi, A.; Loreto, C.; Graziano, A.C.; Cardile, V.; Basile, M.S.; Candido, S.; Falzone, L.; Spandidos, D.A.; et al. Fluoro-edenite induces fibulin-3 overexpression in non-malignant human mesothelial cells. Oncol. Lett. 2016, 12, 3363–3367. [Google Scholar] [CrossRef][Green Version]
- Falzone, L.; Marconi, A.; Loreto, C.; Franco, S.; Spandidos, D.A.; Libra, M. Occupational exposure to carcinogens: Benzene, pesticides and fibers (Review). Mol. Med. Rep. 2016, 14, 4467–4474. [Google Scholar] [CrossRef]
- Malfa, G.A.; Tomasello, B.; Sinatra, F.; Villaggio, G.; Amenta, F.; Avola, R.; Renis, M. “Reactive” response evaluation of primary human astrocytes after methylmercury exposure. J. Neurosci. Res. 2014, 92, 95–103. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef]
- Vivarelli, S.; Falzone, L.; Basile, M.S.; Nicolosi, D.; Genovese, C.; Libra, M.; Salmeri, M. Benefits of using probiotics as adjuvants in anticancer therapy (Review). World Ac. Sci. J. 2019, 1, 125–135. [Google Scholar] [CrossRef]
- Garozzo, A.; Falzone, L.; Rapisarda, V.; Marconi, A.; Cinà, D.; Fenga, C.; Spandidos, D.A.; Libra, M. The risk of HCV infection among health-care workers and its association with extrahepatic manifestations (Review). Mol. Med. Rep. 2017, 15, 3336–3339. [Google Scholar] [CrossRef][Green Version]
- Banna, G.L.; Torino, F.; Marletta, F.; Santagati, M.; Salemi, R.; Cannarozzo, E.; Falzone, L.; Ferraù, F.; Libra, M. Lactobacillus rhamnosus GG: An Overview to Explore the Rationale of Its Use in Cancer. Front. Pharmacol. 2017, 8, 603. [Google Scholar] [CrossRef]
- Margina, D.; Nițulescu, G.; Ungurianu, A.; Mesnage, R.; Goumenou, M.; Sarigiannis, D.; Aschner, M.; Spandidos, D.; Renieri, E.; Hernández, A.; et al. Overview of the effects of chemical mixtures with endocrine disrupting activity in the context of real-life risk simulation (RLRS): An integrative approach (Review). World Ac. Sci. J. 2019, 1, 157–164. [Google Scholar] [CrossRef]
- Tsatsakis, A.M.; Docea, A.O.; Tsitsimpikou, C. New challenges in risk assessment of chemicals when simulating real exposure scenarios; simultaneous multi-chemicals’ low dose exposure. Food Chem. Toxicol. 2016, 96, 174–176. [Google Scholar] [CrossRef]
- Tsatsakis, A.M.; Kouretas, D.; Tzatzarakis, M.N.; Stivaktakis, P.; Tsarouhas, K.; Golokhvast, K.S.; Rakitskii, V.N.; Tutelyan, V.A.; Hernandez, A.F.; Rezaee, R.; et al. Simulating real-life exposures to uncover possible risks to human health: A proposed consensus for a novel methodological approach. Hum. Exp. Toxicol. 2017, 36, 554–564. [Google Scholar] [CrossRef]
- Tsatsakis, A.M.; Docea, A.O.; Calina, D.; Buga, A.M.; Zlatian, O.; Gutnikov, S.; Kostoff, R.N.; Aschner, M. Hormetic Neurobehavioral effects of low dose toxic chemical mixtures in real-life risk simulation (RLRS) in rats. Food Chem. Toxicol. 2019, 125, 141–149. [Google Scholar] [CrossRef]
- Docea, A.O.; Goumenou, M.; Calina, D.; Arsene, A.L.; Dragoi, C.M.; Gofita, E.; Pisoschi, C.G.; Zlatian, O.; Stivaktakis, P.D.; Nikolouzakis, T.K.; et al. Adverse and hormetic effects in rats exposed for 12 months to low dose mixture of 13 chemicals: RLRS part III. Toxicol. Lett. 2019, 310, 70–91. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Docea, A.O.; Constantin, C.; Calina, D.; Zlatian, O.; Nikolouzakis, T.K.; Stivaktakis, P.D.; Kalogeraki, A.; Liesivuori, J.; Tzanakakis, G.; et al. Genotoxic, cytotoxic, and cytopathological effects in rats exposed for 18 months to a mixture of 13 chemicals in doses below NOAEL levels. Toxicol. Lett. 2019, 316, 154–170. [Google Scholar] [CrossRef]
- Tsoukalas, D.; Fragkiadaki, P.; Docea, A.O.; Alegakis, A.K.; Sarandi, E.; Thanasoula, M.; Spandidos, D.A.; Tsatsakis, A.; Razgonova, M.P.; Calina, D. Discovery of potent telomerase activators: Unfolding new therapeutic and anti-aging perspectives. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef]
- Greco, F.A.; Hainsworth, J.D. Introduction: Unknown primary cancer. Semin. Oncol. 2009, 36, 6–7. [Google Scholar] [CrossRef]
- Snyder, T.M.; Khush, K.K.; Valantine, H.A.; Quake, S.R. Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. USA 2011, 108, 6229–6234. [Google Scholar] [CrossRef]
- Nizovtseva, E.V.; Clauvelin, N.; Todolli, S.; Polikanov, Y.S.; Kulaeva, O.I.; Wengrzynek, S.; Olson, W.K.; Studitsky, V.M. Nucleosome-free DNA regions differentially affect distant communication in chromatin. Nucleic Acids Res. 2017, 45, 3059–3067. [Google Scholar] [CrossRef]
- Teif, V.B.; Vainshtein, Y.; Caudron-Herger, M.; Mallm, J.P.; Marth, C.; Höfer, T.; Rippe, K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012, 19, 1185–1192. [Google Scholar] [CrossRef]
- Valouev, A.; Johnson, S.M.; Boyd, S.D.; Smith, C.L.; Fire, A.Z.; Sidow, A. Determinants of nucleosome organization in primary human cells. Nature 2011, 474, 516–520. [Google Scholar] [CrossRef]
- Basile, M.S.; Fagone, P.; Mangano, K.; Mammana, S.; Magro, G.; Salvatorelli, L.; Li Destri, G.; La Greca, G.; Nicoletti, F.; Puleo, S.; et al. KCNMA1 Expression is Downregulated in Colorectal Cancer via Epigenetic Mechanisms. Cancers 2019, 11, 245. [Google Scholar] [CrossRef]
- Balas, M.M.; Johnson, A.M. Exploring the mechanisms behind long noncoding RNAs and cancer. Noncoding RNA Res. 2018, 3, 108–117. [Google Scholar] [CrossRef]
- Chakravarthi, B.V.; Nepal, S.; Varambally, S. Genomic and Epigenomic Alterations in Cancer. Am. J. Pathol. 2016, 186, 1724–1735. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- Candido, S.; Lupo, G.; Pennisi, M.; Basile, M.S.; Anfuso, C.D.; Petralia, M.C.; Gattuso, G.; Vivarelli, S.; Spandidos, D.A.; Libra, M.; et al. The analysis of miRNA expression profiling datasets reveals inverse microRNA patterns in glioblastoma and Alzheimer’s disease. Oncol. Rep. 2019, 42, 911–922. [Google Scholar] [CrossRef]
- Falzone, L.; Romano, G.L.; Salemi, R.; Bucolo, C.; Tomasello, B.; Lupo, G.; Anfuso, C.D.; Spandidos, D.A.; Libra, M.; Candido, S. Prognostic significance of deregulated microRNAs in uveal melanomas. Mol. Med. Rep. 2019, 19, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Zhao, Y.; Huang, W.; Gao, Y.; Xu, W.; Tao, J.; Yang, M.; Li, L.; Ping, W.; Shen, H.; et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 2019, 9, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, L.; Zavridou, M.; Kasimir-Bauer, S.; Lianidou, E.S. Liquid biopsy in ovarian cancer: The potential of circulating miRNAs and exosomes. Transl. Res. 2019, 205, 77–91. [Google Scholar] [CrossRef]
- Menschikowski, M.; Jandeck, C.; Friedemann, M.; Nacke, B.; Hantsche, S.; Tiebel, O.; Sukocheva, O.; Hagelgans, A. Identification of rare levels of methylated tumor DNA fragments using an optimized bias based pre-amplification-digital droplet PCR (OBBPA-ddPCR). Oncotarget 2018, 9, 36137–36150. [Google Scholar] [CrossRef]
- Pardini, B.; Sabo, A.A.; Birolo, G.; Calin, G.A. Noncoding RNAs in Extracellular Fluids as Cancer Biomarkers: The New Frontier of Liquid Biopsies. Cancers 2019, 11, 1170. [Google Scholar] [CrossRef]
- Faria, G.; Silva, E.; Da Fonseca, C.; Quirico-Santos, T. Circulating Cell-Free DNA as a Prognostic and Molecular Marker for Patients with Brain Tumors under Perillyl Alcohol-Based Therapy. Int. J. Mol. Sci. 2018, 19, 1610. [Google Scholar] [CrossRef]
- Huang, Z.H.; Li, L.H.; Hua, D. Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Lett. 2006, 243, 64–70. [Google Scholar] [CrossRef]
- Feng, G.; Ye, X.; Fang, F.; Pu, C.; Huang, H.; Li, G. Quantification of plasma cell-free DNA1 in predicting therapeutic efficacy of sorafenib on metastatic clear cell renal cell carcinoma. Dis. Markers 2013, 34, 105–111. [Google Scholar] [CrossRef]
- Wan, J.; Zhu, L.; Jiang, Z.; Cheng, K. Monitoring of plasma cell-free DNA in predicting postoperative recurrence of clear cell renal cell carcinoma. Urol. Int. 2013, 91, 273–278. [Google Scholar] [CrossRef]
- Madic, J.; Kiialainen, A.; Bidard, F.C.; Birzele, F.; Ramey, G.; Leroy, Q.; Rio Frio, T.; Vaucher, I.; Raynal, V.; Bernard, V.; et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int. J. Cancer 2015, 136, 2158–2165. [Google Scholar] [CrossRef]
- Heida, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16, 421. [Google Scholar] [CrossRef]
- Birkenkamp-Demtröder, K.; Nordentoft, I.; Christensen, E.; Høyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbæk, M.; Jensen, J.B.; Ørntoft, T.F.; et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients With BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Kuang, Y.; O’Connell, A.; Sacher, A.G.; Feeney, N.; Alden, R.S.; Oxnard, G.R.; Paweletz, C.P. Monitoring of Response and Resistance in Plasma of EGFR-Mutant Lung Cancer Using Droplet Digital PCR. Methods Mol. Biol. 2018, 1768, 193–207. [Google Scholar]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef]
- Thompson, J.R.; Menon, S.P. Liquid Biopsies and Cancer Immunotherapy. Cancer J. 2018, 24, 78–83. [Google Scholar] [CrossRef]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Agte, S.; Pan, A.; Simon, N.I.; Iams, W.T.; Cruz, M.R.; Tamragouri, K.; Rhee, K.; Mohindra, N.; et al. Clinical Implications of Circulating Tumor DNA Tumor Mutational Burden (ctDNA TMB) in Non-Small Cell Lung Cancer. Oncologist 2019, 24, 820–828. [Google Scholar] [CrossRef]
- Xi, L.; Pham, T.H.; Payabyab, E.C.; Sherry, R.M.; Rosenberg, S.A.; Raffeld, M. Circulating Tumor DNA as an Early Indicator of Response to T-cell Transfer Immunotherapy in Metastatic Melanoma. Clin. Cancer Res. 2016, 22, 5480–5486. [Google Scholar] [CrossRef]
- Tzanikou, E.; Markou, A.; Politaki, E.; Koutsopoulos, A.; Psyrri, A.; Mavroudis, D.; Georgoulias, V.; Lianidou, E. PIK3CA hotspot mutations in circulating tumor cells and paired circulating tumor DNA in breast cancer: A direct comparison study. Mol. Oncol. 2019. [Google Scholar] [CrossRef]
- Delfau-Larue, M.H.; van der Gucht, A.; Dupuis, J.; Jais, J.P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: Distinct prognostic value in follicular lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef]
- Rossi, G.; Mu, Z.; Rademaker, A.W.; Austin, L.K.; Strickland, K.S.; Costa, R.L.B.; Nagy, R.J.; Zagonel, V.; Taxter, T.J.; Behdad, A.; et al. Cell-Free DNA and Circulating Tumor Cells: Comprehensive Liquid Biopsy Analysis in Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 560–568. [Google Scholar] [CrossRef]
- Leung, F.; Kulasingam, V.; Diamandis, E.P.; Hoon, D.S.; Kinzler, K.; Pantel, K.; Alix-Panabières, C. Circulating Tumor DNA as a Cancer Biomarker: Fact or Fiction? Clin. Chem. 2016, 62, 1054–1060. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Olmedillas-López, S.; García-Arranz, M.; García-Olmo, D. Current and emerging applications of droplet digital PCR in oncology. Mol. Diagn. Ther. 2017, 21, 493–510. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Crona, J.; Verdugo, A.D.; Granberg, D.; Welin, S.; Stålberg, P.; Hellman, P.; Björklund, P. Next-generation sequencing in the clinical genetic screening of patients with pheochromocytoma and paraganglioma. Endocr. Connect. 2013, 2, 104–111. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Bell, A.D.; Usher, C.L.; McCarroll, S.A. Analyzing copy number variation with droplet digital PCR. In Digital PCR; Humana Press: New York, NY, USA, 2018; pp. 143–160. [Google Scholar]
- Karlin-Neumann, G.; Bizouarn, F. Entering the Pantheon of 21 st Century Molecular Biology Tools: A Perspective on Digital PCR. In Digital PCR; Humana Press: New York, NY, USA, 2018; pp. 3–10. [Google Scholar]
- McBride, D.J.; Orpana, A.K.; Sotiriou, C.; Joensuu, H.; Stephens, P.J.; Mudie, L.J.; Hämäläinen, E.; Stebbings, L.A.; Andersson, L.C.; Flanagan, A.M.; et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer 2010, 49, 1062–1069. [Google Scholar] [CrossRef]
- Oellerich, M.; Schütz, E.; Beck, J.; Kanzow, P.; Plowman, P.N.; Weiss, G.J.; Walson, P.D. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit. Rev. Clin. Lab. Sci. 2017, 54, 205–218. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Nordentoft, I.; Høyer, S.; van der Keur, K.; van Kessel, K.; Zwarthoff, E.; Agerbæk, M.; Ørntoft, T.F.; Jensen, J.B.; et al. Liquid Biopsy Analysis of FGFR3 and PIK3CA Hotspot Mutations for Disease Surveillance in Bladder Cancer. Eur. Urol. 2017, 71, 961–969. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget 2017, 8, 14221–14250. [Google Scholar] [CrossRef]
- Falzone, L.; Scola, L.; Zanghì, A.; Biondi, A.; Di Cataldo, A.; Libra, M.; Candido, S. Integrated analysis of colorectal cancer microRNA datasets: Identification of microRNAs associated with tumor development. Aging 2018, 10, 1000–1014. [Google Scholar] [CrossRef]
- Polo, A.; Crispo, A.; Cerino, P.; Falzone, L.; Candido, S.; Giudice, A.; De Petro, G.; Ciliberto, G.; Montella, M.; Budillon, A.; et al. Environment and bladder cancer: Molecular analysis by interaction networks. Oncotarget 2017, 8, 65240–65252. [Google Scholar] [CrossRef]
- Falzone, L.; Candido, S.; Salemi, R.; Basile, M.S.; Scalisi, A.; McCubrey, J.A.; Torino, F.; Signorelli, S.S.; Montella, M.; Libra, M. Computational identification of microRNAs associated to both epithelial to mesenchymal transition and NGAL/MMP-9 pathways in bladder cancer. Oncotarget 2016, 8, 72758–72766. [Google Scholar] [CrossRef]
- Hafsi, S.; Candido, S.; Maestro, R.; Falzone, L.; Soua, Z.; Bonavida, B.; Spandidos, D.A.; Libra, M. Correlation between the overexpression of Yin Yang 1 and the expression levels of miRNAs in Burkitt’s lymphoma: A computational study. Oncol. Lett. 2016, 11, 1021–1025. [Google Scholar] [CrossRef]
- Presti, M.; Mazzon, E.; Basile, M.S.; Petralia, M.C.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Russo, A.; Longo, A.; Avitabile, T.; Nicoletti, F.; Reibaldi, M.; Basile, M.S. Characterization of the Pathophysiological Role of CD47 in Uveal Melanoma. Molecules 2019, 24, 2450. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavò, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NF κB inhibition is associated with OPN/MMP 9 downregulation in cutaneous melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef]
- Fan, G.; Zhang, K.; Ding, J.; Li, J. Prognostic value of EGFR and KRAS in circulating tumor DNA in patients with advanced non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 33922–33932. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Stivaktakis, P.D.; Apalaki, P.; Kalliantasi, K.; Sapsakos, T.M.; Spandidos, D.A.; Tsatsakis, A.; Souglakos, J.; Tsiaoussis, J. Effect of systemic treatment on the micronuclei frequency in the peripheral blood of patients with metastatic colorectal cancer. Oncol. Lett. 2019, 17, 2703–2712. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Popescu, I.D.; Zipeto, D.; Tzanakakis, G.; Nikitovic, D.; Fenga, C.; Stratakis, C.A.; Spandidos, D.A.; Tsatsakis, A.M. Inflammation and Metabolism in Cancer Cell-Mitochondria Key Player. Front. Oncol. 2019, 9, 348. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vassilopoulou, L.; Fragkiadaki, P.; Mariolis Sapsakos, T.; Papadakis, G.Z.; Spandidos, D.A.; Tsatsakis, A.M.; Tsiaoussis, J. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients (Review). Oncol. Rep. 2018, 39, 2455–2472. [Google Scholar] [CrossRef]
- Tänzer, M.; Balluff, B.; Distler, J.; Hale, K.; Leodolter, A.; Röcken, C.; Molnar, B.; Schmid, R.; Lofton-Day, C.; Schuster, T.; et al. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PLoS ONE 2010, 5, e9061. [Google Scholar] [CrossRef]
- Danese, E.; Montagnana, M. Epigenetics of colorectal cancer: Emerging circulating diagnostic and prognostic biomarkers. Ann. Transl. Med. 2017, 5, 279. [Google Scholar] [CrossRef]
- Garrigou, S.; Perkins, G.; Garlan, F.; Normand, C.; Didelot, A.; Le Corre, D.; Peyvandi, S.; Mulot, C.; Niarra, R.; Aucouturier, P.; et al. A Study of Hypermethylated Circulating Tumor DNA as a Universal Colorectal Cancer Biomarker. Clin. Chem. 2016, 62, 1129–1139. [Google Scholar] [CrossRef]
- Jia, S.; Zhang, R.; Li, Z.; Li, J. Clinical and biological significance of circulating tumor cells, circulating tumor DNA, and exosomes as biomarkers in colorectal cancer. Oncotarget 2017, 8, 55632–55645. [Google Scholar] [CrossRef]
- Negrei, C.; Hudita, A.; Ginghina, O.; Galateanu, B.; Voicu, S.N.; Stan, M.; Costache, M.; Fenga, C.; Drakoulis, N.; Tsatsakis, A.M. Colon Cancer Cells Gene Expression Signature As Response to 5- Fluorouracil, Oxaliplatin, and Folinic Acid Treatment. Front. Pharmacol. 2016, 7, 172. [Google Scholar] [CrossRef]
- Takayama, Y.; Suzuki, K.; Muto, Y.; Ichida, K.; Fukui, T.; Kakizawa, N.; Ishikawa, H.; Watanabe, F.; Hasegawa, F.; Saito, M.; et al. Monitoring circulating tumor DNA revealed dynamic changes in KRAS status in patients with metastatic colorectal cancer. Oncotarget 2018, 9, 24398–24413. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Gongora, C.; Guedj, A.S.; Robert, B.; Del Rio, M.; Molina, F.; Lamy, P.J.; Lopez-Crapez, E.; Mathonnet, M.; et al. Circulating Cell-Free DNA from Colorectal Cancer Patients May Reveal High KRAS or BRAF Mutation Load. Transl. Oncol. 2013, 6, 319–328. [Google Scholar] [CrossRef]
- Ghatalia, P.; Smith, C.H.; Winer, A.; Gou, J.; Kiedrowski, L.A.; Slifker, M.; Saltzberg, P.D.; Bubes, N.; Anari, F.M.; Kasireddy, V.; et al. Clinical Utilization Pattern of Liquid Biopsies (LB) to Detect Actionable Driver Mutations, Guide Treatment Decisions and Monitor Disease Burden During Treatment of 33 Metastatic Colorectal Cancer (mCRC) Patients (pts) at a Fox Chase Cancer Center GI Oncology Subspecialty Clinic. Front. Oncol. 2019, 8, 652. [Google Scholar]
- Gabriel, E.; Bagaria, S.P. Assessing the Impact of Circulating Tumor DNA (ctDNA) in Patients with Colorectal Cancer: Separating Fact From Fiction. Front. Oncol. 2018, 8, 297. [Google Scholar] [CrossRef]
- Shang, M.; Chang, C.; Pei, Y.; Guan, Y.; Chang, J.; Li, H. Potential Management of Circulating Tumor DNA as a Biomarker in Triple-Negative Breast Cancer. J. Cancer 2018, 9, 4627–4634. [Google Scholar] [CrossRef]
- Ishiba, T.; Hoffmann, A.C.; Usher, J.; Elshimali, Y.; Sturdevant, T.; Dang, M.; Jaimes, Y.; Tyagi, R.; Gonzales, R.; Grino, M.; et al. Frequencies and expression levels of programmed death ligand 1 (PD-L1) in circulating tumor RNA (ctRNA) in various cancer types. Biochem. Biophys. Res. Commun. 2018, 500, 621–625. [Google Scholar] [CrossRef]
- Kodahl, A.R.; Ehmsen, S.; Pallisgaard, N.; Jylling, A.M.B.; Jensen, J.D.; Laenkholm, A.V.; Knoop, A.S.; Ditzel, H.J. Correlation between circulating cell-free PIK3CA tumor DNA levels and treatment response in patients with PIK3CA-mutated metastatic breast cancer. Mol. Oncol. 2018, 12, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.W.Y.; Murtaza, M.; Wong, A.S.C.; Rueda, O.M.; Smith, C.G.; Chandrananda, D.; Soo, R.A.; Lim, H.L.; Goh, B.C.; Caldas, C.; et al. Dinamics of multiple resistance mechanisms in plasma DNA during EGFR-targeted therapies in non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e7945. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Del Grammastro, M.; Felicioni, L.; Malatesta, S.; Filice, G.; Centi, I.; De Pas, T.; Santoro, A.; Chella, A.; Brandes, A.A.; et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: Toward a real-time liquid biopsy for treatment. PLoS ONE 2014, 9, e103883. [Google Scholar] [CrossRef] [PubMed]
- Boffa, D.J.; Graf, R.P.; Salazar, M.C.; Hoag, J.; Lu, D.; Krupa, R.; Louw, J.; Dugan, L.; Wang, Y.; Landers, M.; et al. Cellular Expression of PD-L1 in the Peripheral Blood of Lung Cancer Patients is Associated with Worse Survival. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 1139–1145. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Raimondi, C.; Mancini, M.; Caponnetto, S.; Gradilone, A.; Gandini, O.; Mastromartino, M.; Del Bene, G.; Prete, A.; Longo, F.; et al. Monitoring PD-L1 positive circulating tumor cells in non-small cell lung cancer patients treated with the PD-1 inhibitor Nivolumab. Sci. Rep. 2016, 6, 31726. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef]
- Myint, N.N.M.; Verma, A.M.; Fernandez-Garcia, D.; Sarmah, P.; Tarpey, P.S.; Al-Aqbi, S.S.; Cai, H.; Trigg, R.; West, K.; Howells, L.M.; et al. Circulating tumor DNA in patients with colorectal adenomas: Assessment of detectability and genetic heterogeneity. Cell Death Dis. 2018, 9, 894. [Google Scholar] [CrossRef]
- Galbiati, S.; Damin, F.; Burgio, V.; Brisci, A.; Soriani, N.; Belcastro, B.; Di Resta, C.; Gianni, L.; Chiari, M.; Ronzoni, M.; et al. Evaluation of three advanced methodologies, COLD-PCR, microarray and ddPCR, for identifying the mutational status by liquid biopsies in metastatic colorectal cancer patients. Clin. Chim. Acta 2019, 489, 136–143. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Takeda, Y.; Wakatsuki, T.; Ichimura, T.; Saiura, A.; Yamaguchi, K.; Takahashi, S.; Noda, T.; Zembutsu, H. Clinical relevance of circulating tumor DNA assessed through deep sequencing in patients with metastatic colorectal cancer. Cancer Med. 2019, 8, 408–417. [Google Scholar] [CrossRef]
- Kondo, Y.; Hayashi, K.; Kawakami, K.; Miwa, Y.; Hayashi, H.; Yamamoto, M. KRAS mutation analysis of single circulating tumor cells from patients with metastatic colorectal cancer. BMC Cancer 2017, 17, 311. [Google Scholar] [CrossRef] [PubMed]
- Damin, F.; Galbiati, S.; Soriani, N.; Burgio, V.; Ronzoni, M.; Ferrari, M.; Chiari, M. Analysis of KRAS, NRAS and BRAF mutational profile by combination of in-tube hybridization and universal tag-microarray in tumor tissue and plasma of colorectal cancer patients. PLoS ONE 2018, 13, e0207876. [Google Scholar] [CrossRef] [PubMed]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef] [PubMed]
- Kidess-Sigal, E.; Liu, H.E.; Triboulet, M.M.; Che, J.; Ramani, V.C.; Visser, B.C.; Poultsides, G.A.; Longacre, T.A.; Marziali, A.; Vysotskaia, V.; et al. Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label-free platform: Comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget 2016, 7, 85349–85364. [Google Scholar] [CrossRef]
- Grützmann, R.; Molnar, B.; Pilarsky, C.; Habermann, J.K.; Schlag, P.M.; Saeger, H.D.; Miehlke, S.; Stolz, T.; Model, F.; Roblick, U.J.; et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS ONE 2008, 3, e3759. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Franovic, A.; Shiotsu, Y.; Kim, S.T.; Kim, K.M.; Banks, K.C.; Raymond, V.M.; Lanman, R.B. Detection of ERBB2 (HER2) Gene Amplification Events in Cell-Free DNA and Response to Anti-HER2 Agents in a Large Asian Cancer Patient Cohort. Front. Oncol. 2019, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, J.; Huang, L.; Chen, L.; Zhou, Y.; Tang, D.; Xie, Y.; Wang, H.; Huang, C. Detection of HER2-positive Circulating Tumor Cells Using the LiquidBiopsy System in Breast Cancer. Clin. Breast Cancer 2019, 19, e239–e246. [Google Scholar] [CrossRef]
- Mazel, M.; Jacot, W.; Pantel, K.; Bartkowiak, K.; Topart, D.; Cayrefourcq, L.; Rossille, D.; Maudelonde, T.; Fest, T.; Alix-Panabières, C. Frequent expression of PD-L1 on circulating breast cancer cells. Mol. Oncol. 2015, 9, 1773–1782. [Google Scholar] [CrossRef]
- Savli, H.; Sertdemir, N.; Aydin, D.; Dursun, B.; Kurtas, O.; Reka, S.; Sunnetci-Akkoyunlu, D.; Eren-Keskin, S.; Uygun, K.; Ozden, E.; et al. TP53, EGFR and PIK3CA gene variations observed as prominent biomarkers in breast and lung cancer by plasma cell-free DNA genomic testing. J. Biotechnol. 2019, 300, 87–93. [Google Scholar] [CrossRef]
- Nakauchi, C.; Kagara, N.; Shimazu, K.; Shimomura, A.; Naoi, Y.; Shimoda, M.; Kim, S.J.; Noguchi, S. Detection of TP53/PIK3CA Mutations in Cell-Free Plasma DNA From Metastatic Breast Cancer Patients Using Next Generation Sequencing. Clin. Breast Cancer 2016, 16, 418–423. [Google Scholar] [CrossRef]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2015, 48, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Ahlborn, L.B.; Tuxen, I.V.; Mouliere, F.; Kinalis, S.; Schmidt, A.Y.; Rohrberg, K.S.; Santoni-Rugiu, E.; Nielsen, F.C.; Lassen, U.; Yde, C.W.; et al. Circulating tumor DNA as a marker of treatment response in BRAF V600E mutated non-melanoma solid tumors. Oncotarget 2018, 9, 32570–32579. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Eder, J.; Pratscher, B.; Brandt, S.; Schneller, D.; Müllegger, R.; Vogl, C.; Trautinger, F.; Brem, G.; Burgstaller, J.P. SNPase-ARMS qPCR: Ultrasensitive Mutation-Based Detection of Cell-Free Tumor DNA in Melanoma Patients. PLoS ONE 2015, 10, e0142273. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Mitra, D.; Sullivan, R.J.; Wittner, B.S.; Kimura, A.M.; Pan, S.; Hoang, M.P.; Brannigan, B.W.; Lawrence, D.P.; Flaherty, K.T.; et al. Isolation and molecular characterization of circulating melanoma cells. Cell Rep. 2014, 7, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, T.C.; Savitch, S.L.; Yee, S.S.; Xu, W.; Huang, A.C.; Harmon, S.; Lieberman, D.B.; Soucier, D.; Fan, R.; Black, T.A.; et al. Feasibility of monitoring advanced melanoma patients using cell-free DNA from plasma. Pigment Cell Melanoma Res. 2018, 31, 73–81. [Google Scholar] [CrossRef]
- Bonaparte, E.; Pesenti, C.; Fontana, L.; Falcone, R.; Paganini, L.; Marzorati, A.; Ferrero, S.; Nosotti, M.; Mendogni, P.; Bareggi, C.; et al. Molecular profiling of lung cancer specimens and liquid biopsies using MALDI-TOF mass spectrometry. Diagn. Pathol. 2018, 13, 4. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939–946. [Google Scholar] [CrossRef]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8. [Google Scholar] [CrossRef]
- Basile, M.S.; Mazzon, E.; Krajnovic, T.; Draca, D.; Cavalli, E.; Al-Abed, Y.; Bramanti, P.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells. Molecules 2018, 23, 2463. [Google Scholar] [CrossRef]
- Chen, J.; Huan, W.; Zuo, H.; Zhao, L.; Huang, C.; Liu, X.; Hou, S.; Qi, J.; Shi, W. Alu methylation serves as a biomarker for non-invasive diagnosis of glioma. Oncotarget 2016, 7, 26099–26106. [Google Scholar] [CrossRef] [PubMed]
- Toraño, E.G.; Petrus, S.; Fernandez, A.F.; Fraga, M.F. Global DNA hypomethylation in cancer: Review of validated methods and clinical significance. Clin. Chem. Lab. Med. 2012, 50, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef] [PubMed]
- Goessl, C.; Müller, M.; Straub, B.; Miller, K. DNA alterations in body fluids as molecular tumor markers for urological malignancies. Eur. Urol. 2002, 41, 668–676. [Google Scholar] [CrossRef]
- Bergerot, P.G.; Hahn, A.W.; Bergerot, C.D.; Jones, J.; Pal, S.K. The Role of Circulating Tumor DNA in Renal Cell Carcinoma. Curr. Treat. Options Oncol. 2018, 19, 10. [Google Scholar] [CrossRef]
- Ulz, P.; Belic, J.; Graf, R.; Auer, M.; Lafer, I.; Fischereder, K.; Webersinke, G.; Pummer, K.; Augustin, H.; Pichler, M.; et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat. Commun. 2016, 7, 12008. [Google Scholar] [CrossRef]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re10. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Parimi, S.; Ko, J.J. Recent advances in circulating tumor cells and cell-free DNA in metastatic prostate cancer: A review. Expert Rev. Anticancer 2017, 17, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Brisuda, A.; Pazourkova, E.; Soukup, V.; Horinek, A.; Hrbáček, J.; Capoun, O.; Svobodova, I.; Pospisilova, S.; Korabecna, M.; Mares, J.; et al. Urinary Cell-Free DNA Quantification as Non-Invasive Biomarker in Patients with Bladder Cancer. Urol. Int. 2016, 96, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Casadio, V.; Calistri, D.; Tebaldi, M.; Bravaccini, S.; Gunelli, R.; Martorana, G.; Bertaccini, A.; Serra, L.; Scarpi, E.; Amadori, D.; et al. Urine cell-free DNA integrity as a marker for early bladder cancer diagnosis: Preliminary data. Urol. Oncol. 2013, 31, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Fantony, J.J.; Longo, T.A.; Gopalakrishna, A.; Owusu, R.; Lance, R.S.; Foo, W.C.; Inman, B.A.; Abern, M.R. Urinary NID2 and TWIST1 methylation to augment conventional urine cytology for the detection of bladder cancer. Cancer Biomark. 2017, 18, 381–387. [Google Scholar] [CrossRef]
- Yegin, Z.; Gunes, S.; Buyukalpelli, R. Hypermethylation of TWIST1 and NID2 in tumor tissues and voided urine in urinary bladder cancer patients. DNA Cell Biol. 2013, 32, 386–392. [Google Scholar] [CrossRef]
- Renard, I.; Joniau, S.; van Cleynenbreugel, B.; Collette, C.; Naômé, C.; Vlassenbroeck, I.; Nicolas, H.; de Leval, J.; Straub, J.; Van Criekinge, W.; et al. Identification and validation of the methylated TWIST1 and NID2 genes through real-time methylation-specific polymerase chain reaction assays for the noninvasive detection of primary bladder cancer in urine samples. Eur. Urol. 2010, 58, 96–104. [Google Scholar] [CrossRef]
- Springer, S.U.; Chen, C.H.; Rodriguez Pena, M.D.C.; Li, L.; Douville, C.; Wang, Y.; Cohen, J.D.; Taheri, D.; Silliman, N.; Schaefer, J.; et al. Non-invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife 2018, 7, e32143. [Google Scholar] [CrossRef]
- Eich, M.L.; Rodriguez Pena, M.D.C.; Springer, S.U.; Taheri, D.; Tregnago, A.C.; Salles, D.C.; Bezerra, S.M.; Cunha, I.W.; Fujita, K.; Ertoy, D.; et al. Incidence and distribution of UroSEEK gene panel in a multi-institutional cohort of bladder urothelial carcinoma. Mod. Pathol. 2019, 32, 1544–1550. [Google Scholar] [CrossRef]
- Haff, L.A.; Belden, A.C.; Hall, L.R.; Ross, P.L.; Smirnov, I.P. SNP Genotyping by MALDI-TOF Mass Spectrometry. In Mass Spectrometry and Genomic Analysis. Housby, J.N., Ed.; Kluwer Academic Publisher: Dordrecht, The Netherlands, 2001; pp. 16–32. [Google Scholar]
- Sharma, V.K.; Vouros, P.; Glick, J. Mass spectrometric based analysis, characterization and applications of circulating cell free DNA isolated from human body fluids. Int. J. Mass Spectrom. 2011, 304, 172–183. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Rink, M.; Shariat, S.F.; Soave, A. Liquid biopsies in bladder cancer—did we find the Holy Grail for biomarker analyses? Transl. Androl. Urol. 2016, 5, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Cirrone, G.A.P.; Margarone, D.; Maggiore, M.; Anzalone, A.; Borghesi, M.; Jia, S.B.; Bulanov, S.S.; Bulanov, S.; Carpinelli, M.; Cavallaro, S.; et al. ELIMED: A new hadron therapy concept based on laser driven ion beams. Proc. SPIE 2013, 8779, 87791. [Google Scholar]
- Van Soest, R.J. Liquid biopsies and plasma DNA: Paving the way for personalized medicine in metastatic castration-resistant prostate cancer. Ann. Oncol. 2017, 28, 1408–1409. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.; Mok, T.S.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, K.Z.; Hui, B.G.; Zhang, K.; Yang, F.; Wang, J. Role of circulating tumor DNA in the management of early-stage lung cancer. Thorac. Cancer 2018, 9, 509–515. [Google Scholar] [CrossRef]
- Babayan, A.; Pantel, K. Advances in liquid biopsy approaches for early detection and monitoring of cancer. Genome Med. 2018, 10, 21. [Google Scholar] [CrossRef]
- Liang, W.; Zhao, Y.; Huang, W.; Liang, H.; Zeng, H.; He, J. Liquid biopsy for early stage lung cancer. J. Thorac. Dis. 2018, 10, S876–S881. [Google Scholar] [CrossRef]
- Krug, A.K.; Enderle, D.; Karlovich, C.; Priewasser, T.; Bentink, S.; Spiel, A.; Brinkmann, K.; Emenegger, J.; Grimm, D.G.; Castellanos-Rizaldos, E.; et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018, 29, 2143. [Google Scholar] [CrossRef]
- Mone, A. Johns Hopkins researchers develop single blood test that screens for eight common cancers. Available online: https://hub.jhu.edu/2018/01/19/cancer-blood-test-johns-hopkins/ (accessed on 13 October 2019).



{kind=link}
{kind=link}
| Characteristics and Scope | Liquid Biopsy | Tissue Biopsy |
|---|---|---|
| Invasiveness | Minimally invasive | Invasive |
| Study prescription time | On demand repeatedly | Prior a therapy prescription |
| Sample degradation | No, long keeping in −70 °C [35] | Cross-linking and DNA fragmentation [35] |
| Tumor size/number of tumor cells for detectable ctDNA | 5 × 107 cells [9] | >109 cells [9] |
| Amount of biomaterial | 3 mL peripheral venous blood | Depending on the technique and organ |
| Screening | Yes [36] | No |
| Therapy choice | Yes | Yes |
| Continuous dynamic observation (monitoring) | Yes, half-life of ctDNA between 16 min and 13 h [32,33,34] | No, too traumatic |
| Response to therapy | Yes [21,34] | No |
| Residual tumors | Yes [21,34] | No |
| Relapse prognosis | Yes [21,34] | No |
| Purpose of Research | Method | Example of Application | Specimen | Sensitivity Specificity % |
|---|---|---|---|---|
| Early diagnostics, screening | Target Deep Sequencing -TamSeq | Ovaries cancer (EGFR, TP53 *) [45] | Plasma | >97/>97 |
| Target Deep Sequencing -Ion-AmpliSeq (Ion Torrent) | Breast cancer (p53, PIK3CA, PTEN, AKT1, IDH2, SMAD4) [27] | Plasma | n/a | |
| Target Deep Sequencing by Illumina- Hi-Seq | Pancreatobiliary Carcinomas (KRAS, TP53, APC, FBXW7, SMAD4) [46] | Plasma | >92/100 | |
| Massively Parallel Sequencing - CancerSEEK | Eight types of cancer (ovaries, liver, stomach, pancreas, esophagus, rectum, lungs and breast) [36] | Plasma | On average 70/>99 | |
| Quantitative Methylation Specific PCR | BC (POU4F2 и PCDH17)7; BC (TWIST1 и NID2) [47,48,49] | Urine | 90/94 90/93 | |
| The liquid typing on microspheres | Gliomas WHO I-IV (The level of Alu methylation) [50] | Plasma | n/a | |
| HPLC ESI-MS-SOMA | HCC (p53) [51] | Plasma | n/a | |
| Identification of cancers of unknown primary | Target Deep Sequencing by Illumina- Hi-Seq or Next-Seq | SCLC, squamous cell lung cancer, colorectal adenocarcinoma, HCC and duct carcinoma of the mammary gland in situ [22] | Plasma | n/a |
| Detection of minimal residual tumor | ddPCR-CNVs | Breast cancer (USP17L2 (DUB3), BRF1, MTA1, and JAG2) [52] | Plasma | n/a |
| Metastasis detection | ddPCR-CNVs | Breast cancer [53] Gastric cancer (MET, HER2) [54,55] | Plasma | 93/100 73.3/93.3 |
| Target Deep Sequencing -TamSeq | Breast cancer (PIK3CA, TP53) [37] | Plasma | n/a | |
| Integral tumor profiling in each specific patient | Massively Parallel Sequencing-PARE + PCR | Specific somatic rearrangements in the chromosomal DNA of solid tumors and plasma ** [56,57] | Tumor tissue + Plasma | n/a |
| Target Deep Sequencing - CAPP-Seq | NSCLC [25,58] | Plasma | 85/96 *** | |
| MALDI-TOF-MS | NSCLC (EGFR, KRAS, BRAF, ALK, PIK3CA, ERBB2, DDR2, AKT, and MEK1) [59] | Tumor tissue + Plasma | n/a | |
| Monitoring of therapy effectiveness and clinical prognosis | Target Deep Sequencing -Tam-Seq and genome-wide sequencing | Invasive bladder muscle cancer [60] | Urine | 83/100 |
| Diagnostics, screening, monitoring etc | ddPCR-SNP and chromosome rearrangement | Different tumor types (PIK3CA KRAS, BRAF, NRAS, and EGFR) ** [31,61,62] | Plasma | 87.2/99.2 |
| Molecular Target | Sample Type | Technology | Study(ies) |
|---|---|---|---|
| Non-Small Cell Lung Carcinoma | |||
| EGFR mutation | ctDNA | ddPCR | [177] |
| ctDNA | ddPCR | [178] | |
| CTCs | NGS | [179] | |
| PD-L1 expression | CTCs | Immunofluorescence | [180] |
| ctRNA | qPCR | [175] | |
| CTCs | Immunofluorescence | [181] | |
| Colorectal Cancer | |||
| APC mutation | ctDNA | NGS | [172] |
| CTCs | NGS | [182] | |
| ctDNA | ddPCR | [183] | |
| KRAS mutation | ctDNA | COLD PCR, Microarray, ddPCR | [184] |
| ctDNA | NGS | [185] | |
| ctDNA | ddPCR | [139] | |
| CTCs | Nested PCR | [186] | |
| BRAF mutation | ctDNA | PCR-Microarray | [187] |
| ctDNA | ddPCR | [188] | |
| CTCs | qPCR | [189] | |
| mSEPT9 methylation | ctDNA | Real-Time PCR | [190] |
| Breast Cancer | |||
| HER2 expression | ctDNA | NGS | [191] |
| CTCs | Immunofluorescence | [192] | |
| PD-L1 expression | ctRNA | qPCR | [175] |
| CTCs | Western blot Flow Cytometry Immunocytochemistry | [193] | |
| PIK3CA mutation | ctDNA | ddPCR | [176] |
| ctDNA | NGS | [194] | |
| ctDNA | NGS | [195] | |
| CTCs | ddPCR | [134] | |
| Cutaneous Melanoma | |||
| BRAF mutation | ctDNA | ddPCR | [126] |
| CTCs | ddPCR | [196] | |
| ctDNA | Exome NGS | [197] | |
| PTEN mutation | ctDNA | SNPase-ARMS qPCR | [198] |
| CTCs | NGS RNA | [199] | |
| TERT promoter mutation | ctDNA | ddPCR | [200] |
| KIT mutation | ctDNA | NGS | [201] |
| CTCs | hemi-nested PCR | [202] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuaeva, N.O.; Falzone, L.; Porozov, Y.B.; Nosyrev, A.E.; Trukhan, V.M.; Kovatsi, L.; Spandidos, D.A.; Drakoulis, N.; Kalogeraki, A.; Mamoulakis, C.; et al. Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements. Cells 2019, 8, 1251. https://doi.org/10.3390/cells8101251
Tuaeva NO, Falzone L, Porozov YB, Nosyrev AE, Trukhan VM, Kovatsi L, Spandidos DA, Drakoulis N, Kalogeraki A, Mamoulakis C, et al. Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements. Cells. 2019; 8(10):1251. https://doi.org/10.3390/cells8101251
Chicago/Turabian StyleTuaeva, Natalia O., Luca Falzone, Yuri B. Porozov, Alexander E. Nosyrev, Vladimir M. Trukhan, Leda Kovatsi, Demetrios A. Spandidos, Nikolaos Drakoulis, Alexandra Kalogeraki, Charalampos Mamoulakis, and et al. 2019. "Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements" Cells 8, no. 10: 1251. https://doi.org/10.3390/cells8101251
APA StyleTuaeva, N. O., Falzone, L., Porozov, Y. B., Nosyrev, A. E., Trukhan, V. M., Kovatsi, L., Spandidos, D. A., Drakoulis, N., Kalogeraki, A., Mamoulakis, C., Tzanakakis, G., Libra, M., & Tsatsakis, A. (2019). Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements. Cells, 8(10), 1251. https://doi.org/10.3390/cells8101251