The Glucocorticoid Receptor in Cardiovascular Health and Disease


Abstract
1. Introduction
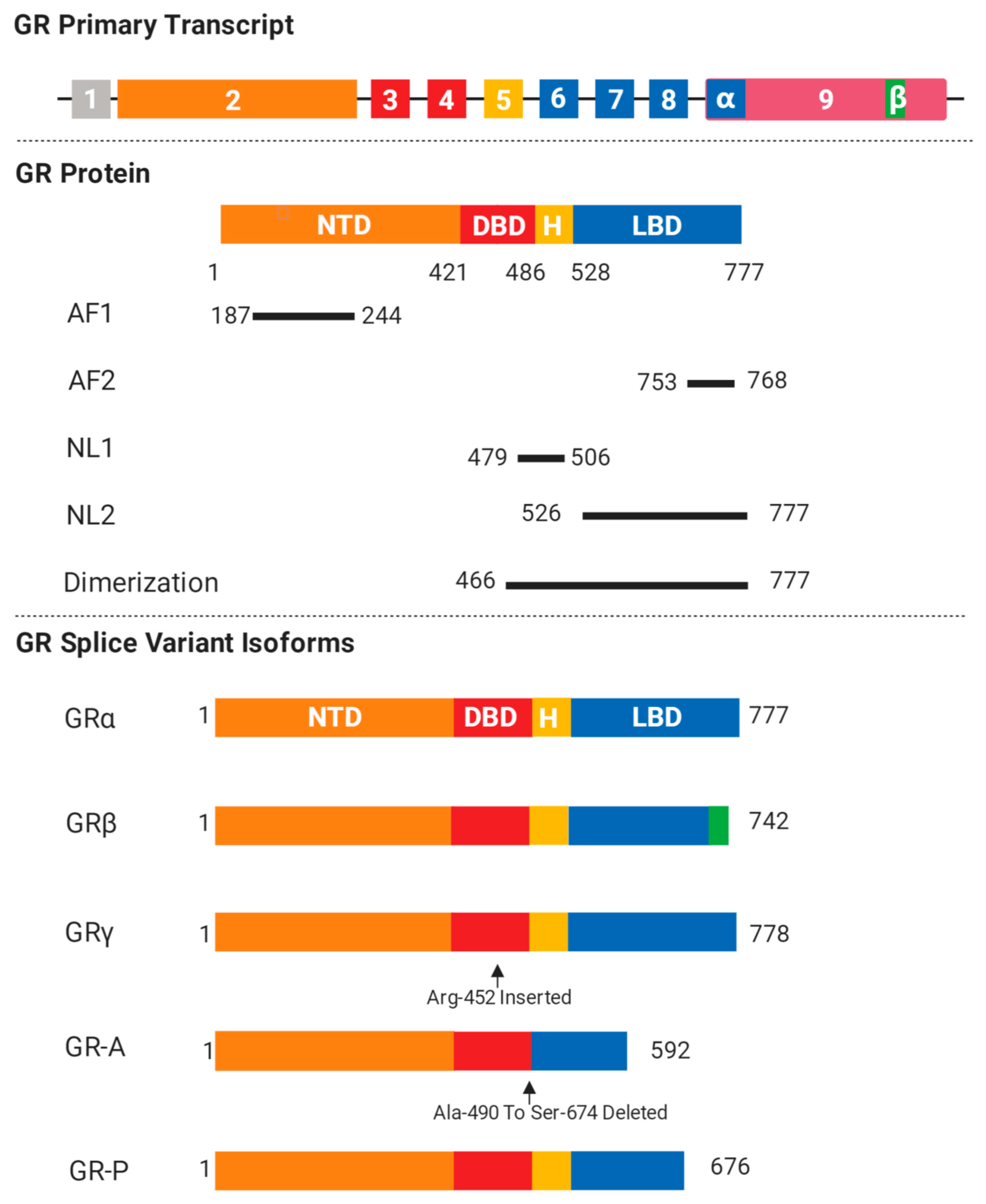
2. GR Isoforms and Structure
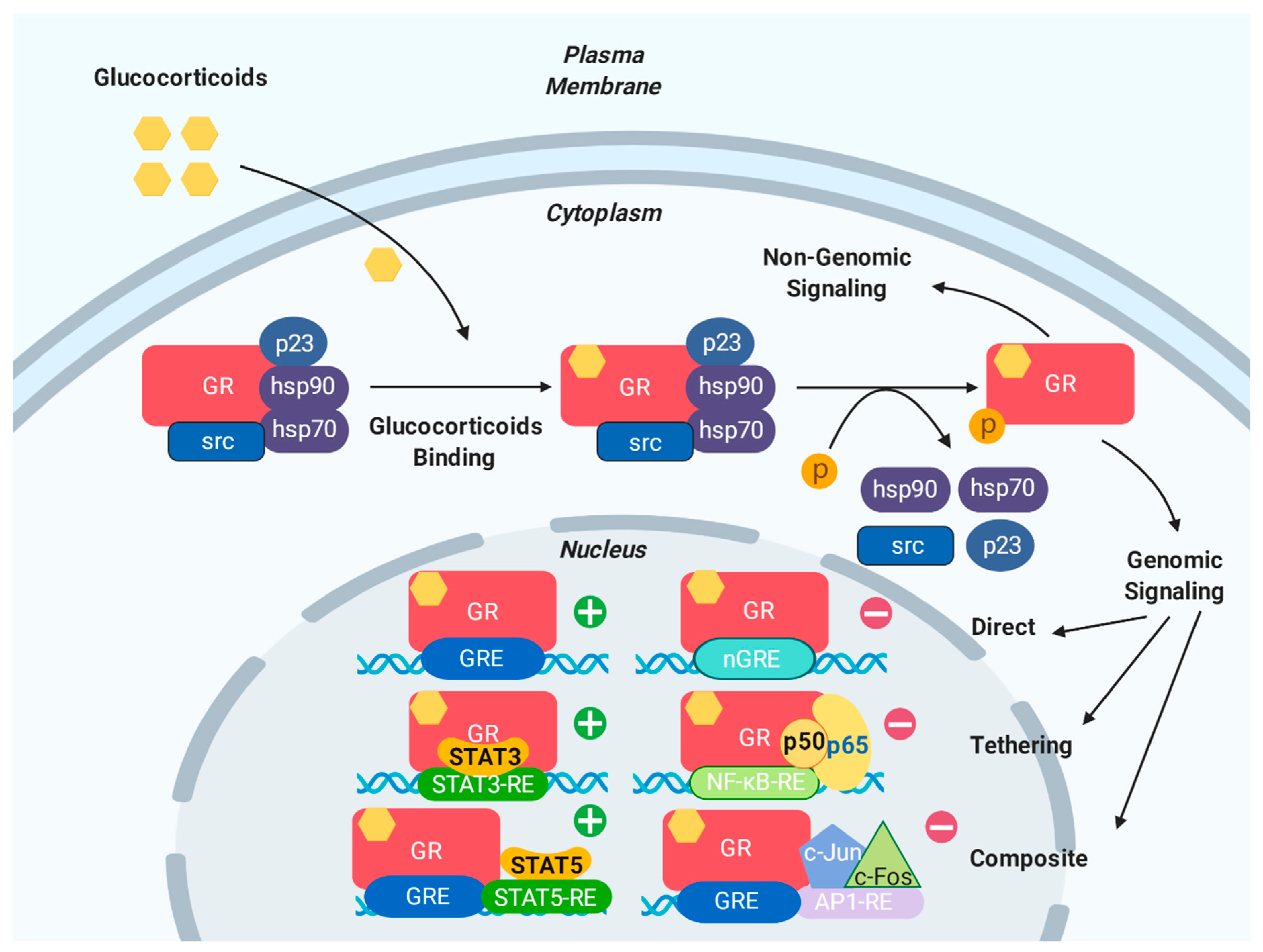
3. Genomic and Non-Genomic Effects of GR
3.1. Genomic Effects of GR
3.2. Non-Genomic Effects of GR
4. Glucocorticoid Metabolism in the Cardiovascular System
5. GR Polymorphisms
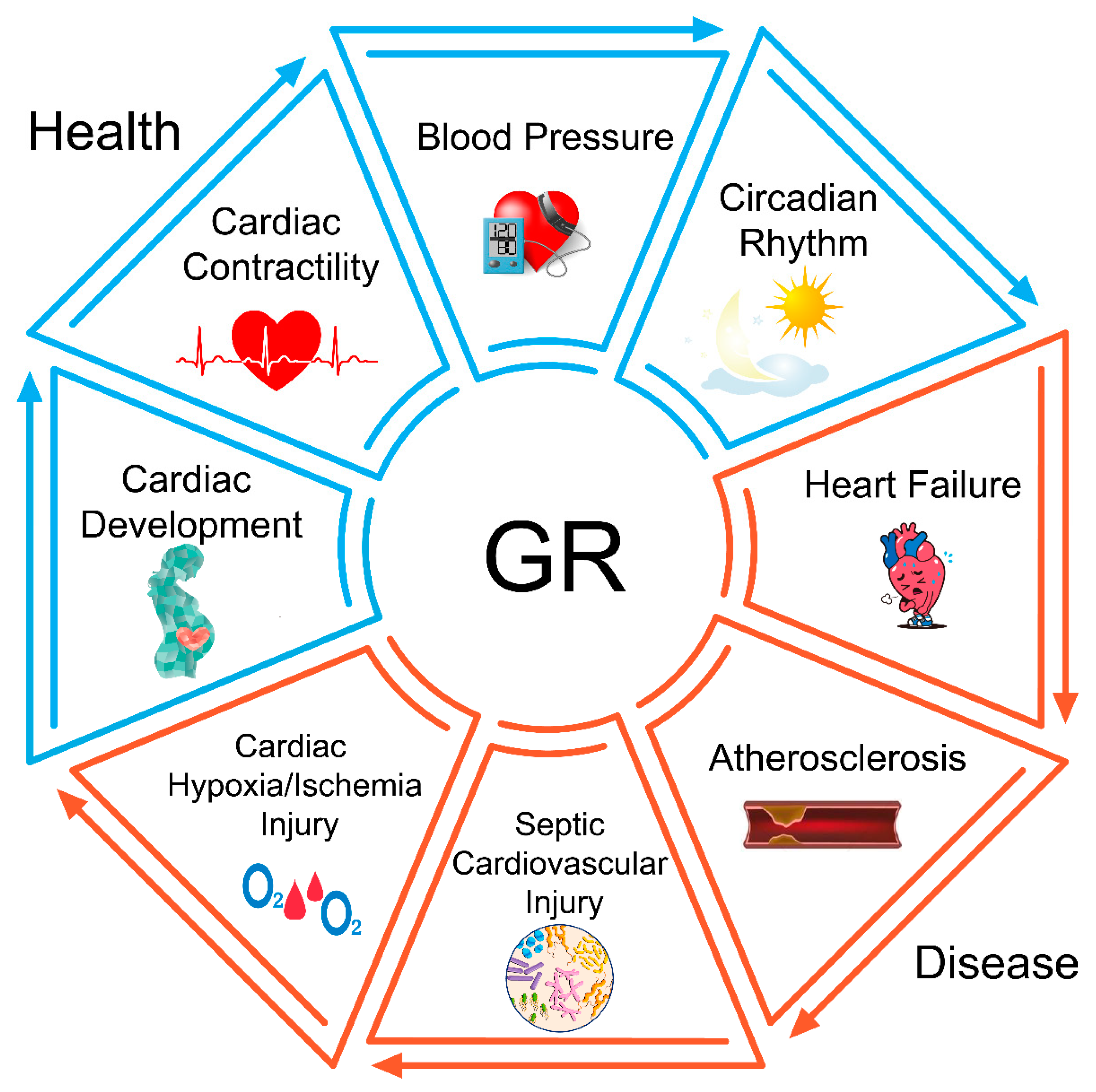
6. The Role of GR in Physiological Conditions
6.1. GR in Cardiac Development
6.2. GR in Cardiac Contractility
6.3. GR in Blood Pressure Regulation
6.4. GR in the Circadian Rhythm
7. The Role of GR in Cardiovascular Diseases
7.1. GR in Heart Failure
7.2. GR in Atherosclerosis
7.3. GR in Sepsis-Induced Cardiovascular Injury
7.4. GR in Cardiac Hypoxia/Ischemia Injury
8. Conclusions
Future Directions
- Targeting endothelial GR-linked pathways in septic shock or atherosclerosis.
- Modulating cardiomyocyte GR-linked pathways in heart failure or myocardial infarction
- Investigating GR regulation of peripheral circadian rhythms.
- Controlling tissue microenvironments by 11-β-HSDs.
- Exploring cardiovascular risk reduction models using GR biology.
Funding
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 8 October 2019).
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the global burden of cardiovascular disease, part 1: The epidemiology and risk factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.E.; Poltavskiy, E.; Janak, J.C.; Beyer, C.A.; Stewart, I.J.; Howard, J.T. Us military service and racial/ethnic differences in cardiovascular disease: An analysis of the 2011–2016 behavioral risk factor surveillance system. Ethn. Dis. 2019, 29, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Maharani, A.; Sujarwoto; Praveen, D.; Oceady, D.; Tampubolon, G.; Patel, A. Cardiovascular disease risk factor prevalence and estimated 10-year cardiovascular risk scores in indonesia: The smarthealth extend study. PLoS ONE 2019, 14, e0215219. [Google Scholar] [CrossRef] [PubMed]
- Kashani, M.; Eliasson, A.; Vernalis, M.; Costa, L.; Terhaar, M. Improving assessment of cardiovascular disease risk by using family history: An integrative literature review. J. Cardiovasc. Nurs. 2013, 28, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid signaling in health and disease: Insights from tissue-specific gr knockout mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure:Function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. Editorial: Cardiac steroidogenesis--new sites of synthesis, or much ado about nothing? J. Clin. Endocrinol. Metab. 2001, 86, 5118–5120. [Google Scholar]
- Lee, S.R.; Kim, H.K.; Youm, J.B.; Dizon, L.A.; Song, I.S.; Jeong, S.H.; Seo, D.Y.; Ko, K.S.; Rhee, B.D.; Kim, N.; et al. Non-genomic effect of glucocorticoids on cardiovascular system. Pflug. Arch. 2012, 464, 549–559. [Google Scholar] [CrossRef]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, 11–24. [Google Scholar] [CrossRef]
- Nussinovitch, U.; de Carvalho, J.F.; Pereira, R.M.; Shoenfeld, Y. Glucocorticoids and the cardiovascular system: State of the art. Curr. Pharm. Des. 2010, 16, 3574–3585. [Google Scholar] [CrossRef]
- Zhou, J.; Cidlowski, J.A. The human glucocorticoid receptor: One gene, multiple proteins and diverse responses. Steroids 2005, 70, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Structure and function of steroid receptor af1 transactivation domains: Induction of active conformations. Biochem. J. 2005, 391, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Cellular processing of the glucocorticoid receptor gene and protein: New mechanisms for generating tissue-specific actions of glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. Translational regulatory mechanisms generate n-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell. 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Collins, J.B.; Grissom, S.F.; Cidlowski, J.A. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol. Cell. Biol. 2007, 27, 7143–7160. [Google Scholar] [CrossRef]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. 2009, 66, 3435–3448. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Cidlowski, J.A. The physiology of human glucocorticoid receptor beta (hgrbeta) and glucocorticoid resistance. Ann. N. Y. Acad. Sci. 2006, 1069, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Bowen, H.; Jee, Y.K.; Mahfiche, N.; Soh, C.; Lee, T.; Hawrylowicz, C.; Lavender, P. The glucocorticoid receptor beta isoform can mediate transcriptional repression by recruiting histone deacetylases. J. Allergy Clin. Immunol. 2008, 121, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Manoli, I.; Kelkar, S.; Wang, Y.; Su, Y.A.; Chrousos, G.P. Glucocorticoid receptor (gr) beta has intrinsic, gralpha-independent transcriptional activity. Biochem. Biophys. Res. Commun. 2009, 381, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Beger, C.; Gerdes, K.; Lauten, M.; Tissing, W.J.; Fernandez-Munoz, I.; Schrappe, M.; Welte, K. Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br. J. Haematol. 2003, 122, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.W.; Davis, J.R.; White, A.; Clark, A.J. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer Res. 1996, 56, 3276–3280. [Google Scholar] [PubMed]
- Moalli, P.A.; Pillay, S.; Krett, N.L.; Rosen, S.T. Alternatively spliced glucocorticoid receptor messenger rnas in glucocorticoid-resistant human multiple myeloma cells. Cancer Res. 1993, 53, 3877–3879. [Google Scholar]
- de Lange, P.; Segeren, C.M.; Koper, J.W.; Wiemer, E.; Sonneveld, P.; Brinkmann, A.O.; White, A.; Brogan, I.J.; de Jong, F.H.; Lamberts, S.W. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res. 2001, 61, 3937–3941. [Google Scholar]
- Gaitan, D.; DeBold, C.R.; Turney, M.K.; Zhou, P.; Orth, D.N.; Kovacs, W.J. Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Mol. Endocrinol. 1995, 9, 1193–1201. [Google Scholar]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.; Hubner, N.; Evans, R.M. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol. Cell. 2013, 49, 158–171. [Google Scholar] [CrossRef]
- Hudson, W.H.; Youn, C.; Ortlund, E.A. The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 2013, 20, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [PubMed]
- Whitworth, J.A. Studies on the mechanisms of glucocorticoid hypertension in humans. Blood Press 1994, 3, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, W.W.; Jansen, P.M.; van den Bogaerdt, A.J.; AH, J.D. Angiotensin ii-aldosterone interaction in human coronary microarteries involves gpr30, egfr, and endothelial no synthase. Cardiovasc. Res. 2012, 94, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Shaltout, H.A.; Rose, J.C.; Figueroa, J.P.; Chappell, M.C.; Diz, D.I.; Averill, D.B. Acute at(1)-receptor blockade reverses the hemodynamic and baroreflex impairment in adult sheep exposed to antenatal betamethasone. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, H. Pathophysiological role of angiotensin ii type 2 receptor in cardiovascular and renal diseases. Circ. Res. 1998, 83, 1182–1191. [Google Scholar] [CrossRef]
- Talaber, G.; Boldizsar, F.; Bartis, D.; Palinkas, L.; Szabo, M.; Berta, G.; Setalo, G., Jr.; Nemeth, P.; Berki, T. Mitochondrial translocation of the glucocorticoid receptor in double-positive thymocytes correlates with their sensitivity to glucocorticoid-induced apoptosis. Int. Immunol. 2009, 21, 1269–1276. [Google Scholar] [CrossRef]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef]
- Buttgereit, F.; Straub, R.H.; Wehling, M.; Burmester, G.R. Glucocorticoids in the treatment of rheumatic diseases: An update on the mechanisms of action. Arthritis. Rheum. 2004, 50, 3408–3417. [Google Scholar] [CrossRef]
- Hedman, E.; Widen, C.; Asadi, A.; Dinnetz, I.; Schroder, W.P.; Gustafsson, J.A.; Wikstrom, A.C. Proteomic identification of glucocorticoid receptor interacting proteins. Proteomics 2006, 6, 3114–3126. [Google Scholar] [CrossRef]
- McMaster, A.; Ray, D.W. Modelling the glucocorticoid receptor and producing therapeutic agents with anti-inflammatory effects but reduced side-effects. Exp. Physiol. 2007, 92, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Urban, G.; Scammell, J.G.; Dean, N.M.; McLean, T.K.; Aragon, I.; Honkanen, R.E. Ser/thr protein phosphatase type 5 (pp5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry 1999, 38, 8849–8857. [Google Scholar] [CrossRef] [PubMed]
- Kfir-Erenfeld, S.; Sionov, R.V.; Spokoini, R.; Cohen, O.; Yefenof, E. Protein kinase networks regulating glucocorticoid-induced apoptosis of hematopoietic cancer cells: Fundamental aspects and practical considerations. Leuk. Lymphoma 2010, 51, 1968–2005. [Google Scholar] [CrossRef] [PubMed]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of action in health and disease. Rheum. Dis. Clin. North. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.G.; Bagley, C.J.; Elder, P.A.; Bachmann, A.W.; Torpy, D.J. Plasma free cortisol fraction reflects levels of functioning corticosteroid-binding globulin. Clin. Chim. Acta 2005, 359, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Gross, K.L.; Cidlowski, J.A. Tissue-specific glucocorticoid action: A family affair. Trends Endocrinol. Metab. 2008, 19, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kino, T. Single nucleotide variations of the human gr gene manifested as pathologic mutations or polymorphisms. Endocrinology 2018, 159, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, E.F.; Koper, J.W.; Huizenga, N.A.; Uitterlinden, A.G.; Janssen, J.A.; Brinkmann, A.O.; Grobbee, D.E.; de Jong, F.H.; van Duyn, C.M.; Pols, H.A.; et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes 2002, 51, 3128–3134. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, A.M.; van Rossum, E.F.; Maestrini, S.; Berselli, M.E.; Tagliaferri, M.; Podesta, F.; Koper, J.W.; Liuzzi, A.; Lamberts, S.W. The relation between two polymorphisms in the glucocorticoid receptor gene and body mass index, blood pressure and cholesterol in obese patients. Clin. Endocrinol. 2003, 59, 68–74. [Google Scholar] [CrossRef]
- Cellini, E.; Castellini, G.; Ricca, V.; Bagnoli, S.; Tedde, A.; Rotella, C.M.; Faravelli, C.; Sorbi, S.; Nacmias, B. Glucocorticoid receptor gene polymorphisms in italian patients with eating disorders and obesity. Psychiatr. Genet. 2010, 20, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Wang, X.L.; Dalziel, B.; Caterson, I.D.; Morris, B.J. Association of obesity, but not diabetes or hypertension, with glucocorticoid receptor n363s variant. Obes. Res. 2003, 11, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Roussel, R.; Reis, A.F.; Dubois-Laforgue, D.; Bellanne-Chantelot, C.; Timsit, J.; Velho, G. The n363s polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin. Endocrinol. Oxf. 2003, 59, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Wang, X.L.; Morris, B.J. Association of coronary artery disease with glucocorticoid receptor n363s variant. Hypertension 2003, 41, 404–407. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geelhoed, J.J.; van Duijn, C.; van Osch-Gevers, L.; Steegers, E.A.; Hofman, A.; Helbing, W.A.; Jaddoe, V.W. Glucocorticoid receptor-9beta polymorphism is associated with systolic blood pressure and heart growth during early childhood. The generation r study. Early Hum. Dev. 2011, 87, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Cuzzoni, E.; De Iudicibus, S.; Bartoli, F.; Ventura, A.; Decorti, G. Association between bcli polymorphism in the nr3c1 gene and in vitro individual variations in lymphocyte responses to methylprednisolone. Br. J. Clin. Pharmacol. 2012, 73, 651–655. [Google Scholar] [CrossRef]
- van Rossum, E.F.; Koper, J.W.; van den Beld, A.W.; Uitterlinden, A.G.; Arp, P.; Ester, W.; Janssen, J.A.; Brinkmann, A.O.; de Jong, F.H.; Grobbee, D.E.; et al. Identification of the bcli polymorphism in the glucocorticoid receptor gene: Association with sensitivity to glucocorticoids in vivo and body mass index. Clin. Endocrinol. Oxf. 2003, 59, 585–592. [Google Scholar] [CrossRef]
- Giordano, R.; Marzotti, S.; Berardelli, R.; Karamouzis, I.; Brozzetti, A.; D’Angelo, V.; Mengozzi, G.; Mandrile, G.; Giachino, D.; Migliaretti, G.; et al. Bcli polymorphism of the glucocorticoid receptor gene is associated with increased obesity, impaired glucose metabolism and dyslipidaemia in patients with addison’s disease. Clin. Endocrinol. Oxf. 2012, 77, 863–870. [Google Scholar] [CrossRef]
- Rosmond, R.; Chagnon, Y.C.; Holm, G.; Chagnon, M.; Perusse, L.; Lindell, K.; Carlsson, B.; Bouchard, C.; Bjorntorp, P. A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic-pituitary-adrenal axis. Obes. Res. 2000, 8, 211–218. [Google Scholar] [CrossRef]
- Steiger, H.; Gauvin, L.; Joober, R.; Israel, M.; Badawi, G.; Groleau, P.; Bruce, K.R.; Yin Kin, N.M.; Sycz, L.; Ouelette, A.S. Interaction of the bcii glucocorticoid receptor polymorphism and childhood abuse in bulimia nervosa (bn): Relationship to bn and to associated trait manifestations. J. Psychiatr. Res. 2012, 46, 152–158. [Google Scholar] [CrossRef]
- Hauer, D.; Weis, F.; Papassotiropoulos, A.; Schmoeckel, M.; Beiras-Fernandez, A.; Lieke, J.; Kaufmann, I.; Kirchhoff, F.; Vogeser, M.; Roozendaal, B.; et al. Relationship of a common polymorphism of the glucocorticoid receptor gene to traumatic memories and posttraumatic stress disorder in patients after intensive care therapy. Crit. Care Med. 2011, 39, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Koeijvoets, K.C.; van der Net, J.B.; van Rossum, E.F.; Steyerberg, E.W.; Defesche, J.C.; Kastelein, J.J.; Lamberts, S.W.; Sijbrands, E.J. Two common haplotypes of the glucocorticoid receptor gene are associated with increased susceptibility to cardiovascular disease in men with familial hypercholesterolemia. J. Clin. Endocrinol. Metab. 2008, 93, 4902–4908. [Google Scholar] [CrossRef] [PubMed]
- Ukkola, O.; Rosmond, R.; Tremblay, A.; Bouchard, C. Glucocorticoid receptor bcl i variant is associated with an increased atherogenic profile in response to long-term overfeeding. Atherosclerosis 2001, 157, 221–224. [Google Scholar] [CrossRef]
- Charmandari, E.; Ichijo, T.; Jubiz, W.; Baid, S.; Zachman, K.; Chrousos, G.P.; Kino, T. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hgr) gene enhancing hgr-mediated gene expression. J. Clin. Endocrinol. Metab. 2008, 93, 4963–4968. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Bachrach, B.E.; Hurt, D.E.; Gajula, S.; Pittman, A.; Lescher, R.; Kino, T. A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J. Clin. Endocrinol. Metab. 2010, 95, 2281–2285. [Google Scholar] [CrossRef]
- Charmandari, E.; Kino, T.; Ichijo, T.; Jubiz, W.; Mejia, L.; Zachman, K.; Chrousos, G.P. A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J. Clin. Endocrinol. Metab. 2007, 92, 3986–3990. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.K.; Pretorius, C.J.; Ungerer, J.P.; Salmon, N.J.; Conwell, L.S.; Pearen, M.A.; Batch, J.A. Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in helix 12 of the ligand binding domain of the glucocorticoid receptor gene (nr3c1). J. Clin. Endocrinol. Metab. 2010, 95, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Roberts, M.L.; Kino, T.; Braatvedt, G.; Hurt, D.E.; Katsantoni, E.; Sertedaki, A.; Chrousos, G.P.; Charmandari, E. A novel point mutation of the human glucocorticoid receptor gene causes primary generalized glucocorticoid resistance through impaired interaction with the lxxll motif of the p160 coactivators: Dissociation of the transactivating and transreppressive activities. J. Clin. Endocrinol. Metab. 2014, 99, 902–907. [Google Scholar]
- Hurley, D.M.; Accili, D.; Stratakis, C.A.; Karl, M.; Vamvakopoulos, N.; Rorer, E.; Constantine, K.; Taylor, S.I.; Chrousos, G.P. Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J. Clin. Investig. 1991, 87, 680–686. [Google Scholar] [CrossRef]
- Raef, H.; Baitei, E.Y.; Zou, M.; Shi, Y. Genotype-phenotype correlation in a family with primary cortisol resistance: Possible modulating effect of the er22/23ek polymorphism. Eur. J. Endocrinol. 2008, 158, 577–582. [Google Scholar] [CrossRef]
- Bertalan, R.; Patocs, A.; Boyle, B.; Rigo, J.; Racz, K. The protective effect of the er22/23ek polymorphism against an excessive weight gain during pregnancy. Gynecol. Endocrinol. 2009, 25, 379–382. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, E.F.; Lamberts, S.W. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Marti, A.; Ochoa, M.C.; Sanchez-Villegas, A.; Martinez, J.A.; Martinez-Gonzalez, M.A.; Hebebrand, J.; Hinney, A.; Vedder, H. Meta-analysis on the effect of the n363s polymorphism of the glucocorticoid receptor gene (grl) on human obesity. BMC Med. Genet. 2006, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Li, J.; Forhead, A.J. Glucocorticoids and the preparation for life after birth: Are there long-term consequences of the life insurance? Proc. Nutr. Soc. 1998, 57, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Blendy, J.A.; Monaghan, A.P.; Krieglstein, K.; Schmid, W.; Aguzzi, A.; Fantuzzi, G.; Hummler, E.; Unsicker, K.; Schutz, G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995, 9, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3269–3282. [Google Scholar] [CrossRef]
- Oakley, R.H.; Ren, R.; Cruz-Topete, D.; Bird, G.S.; Myers, P.H.; Boyle, M.C.; Schneider, M.D.; Willis, M.S.; Cidlowski, J.A. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. USA 2013, 110, 17035–17040. [Google Scholar] [CrossRef]
- Rog-Zielinska, E.A.; Craig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for pgc-1alpha. Cell. Death. Differ. 2015, 22, 1106–1116. [Google Scholar] [CrossRef]
- Kim, M.Y.; Eiby, Y.A.; Lumbers, E.R.; Wright, L.L.; Gibson, K.J.; Barnett, A.C.; Lingwood, B.E. Effects of glucocorticoid exposure on growth and structural maturation of the heart of the preterm piglet. PLoS ONE 2014, 9, e93407. [Google Scholar] [CrossRef]
- Eiby, Y.A.; Lumbers, E.R.; Headrick, J.P.; Lingwood, B.E. Left ventricular output and aortic blood flow in response to changes in preload and afterload in the preterm piglet heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, 769–777. [Google Scholar] [CrossRef]
- Abbasi, S.; Hirsch, D.; Davis, J.; Tolosa, J.; Stouffer, N.; Debbs, R.; Gerdes, J.S. Effect of single versus multiple courses of antenatal corticosteroids on maternal and neonatal outcome. Am. J. Obstet. Gynecol. 2000, 182, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Crowther, C.A.; Haslam, R.R.; Hiller, J.E.; Doyle, L.W.; Robinson, J.S.; Australasian Collaborative Trial of Repeat Doses of Steroids Study Group. Neonatal respiratory distress syndrome after repeat exposure to antenatal corticosteroids: A randomised controlled trial. Lancet 2006, 367, 1913–1919. [Google Scholar] [CrossRef]
- Gay, M.S.; Li, Y.; Xiong, F.; Lin, T.; Zhang, L. Dexamethasone treatment of newborn rats decreases cardiomyocyte endowment in the developing heart through epigenetic modifications. PLoS ONE 2015, 10, e0125033. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Lin, T.; Song, M.; Ma, Q.; Martinez, S.R.; Lv, J.; MataGreenwood, E.; Xiao, D.; Xu, Z.; Zhang, L. Antenatal hypoxia induces epigenetic repression of glucocorticoid receptor and promotes ischemic-sensitive phenotype in the developing heart. J. Mol. Cell. Cardiol. 2016, 91, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Tymianski, M.; Tator, C.H. Normal and abnormal calcium homeostasis in neurons: A basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 1996, 38, 1176–1195. [Google Scholar] [PubMed]
- Bers, D. Sources and Sinks of Extracellular Calcium. In Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Springer: Dordrecht, The Netherlands, 2001; Volume 237, pp. 39–62. [Google Scholar]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and excitation-contraction coupling in the heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cruz-Topete, D.; He, B.; Foley, J.F.; Myers, P.H.; Xu, X.; Gomez-Sanchez, C.E.; Chambon, P.; Willis, M.S.; Cidlowski, J.A. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci. Signal. 2019, 12, 577. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; Oakley, R.H.; Carroll, N.G.; He, B.; Myers, P.H.; Xu, X.; Watts, M.N.; Trosclair, K.; Glasscock, E.; Dominic, P.; et al. Deletion of the cardiomyocyte glucocorticoid receptor leads to sexually dimorphic changes in cardiac gene expression and progression to heart failure. J. Am. Heart Assoc. 2019, 8, e011012. [Google Scholar] [CrossRef]
- Falcon, D.; Gonzalez-Montelongo, R.; Sanchez de Rojas-de Pedro, E.; Ordonez, A.; Urena, J.; Castellano, A. Dexamethasone-induced upregulation of cav3.2 t-type ca(2+) channels in rat cardiac myocytes. J. Steroid. Biochem. Mol. Biol. 2018, 178, 193–202. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Geller, D.S. Glucocorticoid-induced hypertension. Pediatr. Nephrol. 2012, 27, 1059–1066. [Google Scholar] [CrossRef]
- Provencher, P.H.; Saltis, J.; Funder, J.W. Glucocorticoids but not mineralocorticoids modulate endothelin-1 and angiotensin ii binding in shr vascular smooth muscle cells. J. Steroid. Biochem. Mol. Biol. 1995, 52, 219–225. [Google Scholar] [CrossRef]
- Kornel, L.; Nelson, W.A.; Manisundaram, B.; Chigurupati, R.; Hayashi, T. Mechanism of the effects of glucocorticoids and mineralocorticoids on vascular smooth muscle contractility. Steroids 1993, 58, 580–587. [Google Scholar] [CrossRef]
- Tsugita, M.; Iwasaki, Y.; Nishiyama, M.; Taguchi, T.; Shinahara, M.; Taniguchi, Y.; Kambayashi, M.; Terada, Y.; Hashimoto, K. Differential regulation of 11beta-hydroxysteroid dehydrogenase type-1 and -2 gene transcription by proinflammatory cytokines in vascular smooth muscle cells. Life Sci. 2008, 83, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Wallerath, T.; Witte, K.; Schafer, S.C.; Schwarz, P.M.; Prellwitz, W.; Wohlfart, P.; Kleinert, H.; Lehr, H.A.; Lemmer, B.; Forstermann, U. Down-regulation of the expression of endothelial no synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc. Natl. Acad. Sci. USA 1999, 96, 13357–13362. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.P.; Searle, N. Glucocorticoid inhibition of cytokine-induced e-selectin promoter activation. Biochem. Soc. Trans. 1997, 25, 189. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Zhang, J.; Geller, D.S. A critical role for vascular smooth muscle in acute glucocorticoid-induced hypertension. J. Am. Soc. Nephrol. 2008, 19, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Zhang, J.; Gonzalez, D.; Albinsson, S.; Geller, D.S. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J. Hypertens. 2011, 29, 1347–1356. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef]
- Sato, A.; Suzuki, H.; Nakazato, Y.; Shibata, H.; Inagami, T.; Saruta, T. Increased expression of vascular angiotensin ii type 1a receptor gene in glucocorticoid-induced hypertension. J. Hypertens. 1994, 12, 511–516. [Google Scholar] [CrossRef]
- Kornel, L.; Prancan, A.V.; Kanamarlapudi, N.; Hynes, J.; Kuzianik, E. Study on the mechanisms of glucocorticoid-induced hypertension: Glucocorticoids increase transmembrane ca2+ influx in vascular smooth muscle in vivo. Endocr. Res. 1995, 21, 203–210. [Google Scholar] [CrossRef]
- Mitchell, B.M.; Dorrance, A.M.; Mack, E.A.; Webb, R.C. Glucocorticoids decrease gtp cyclohydrolase and tetrahydrobiopterin-dependent vasorelaxation through glucocorticoid receptors. J. Cardiovasc. Pharmacol. 2004, 43, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Cermakian, N.; Sassone-Corsi, P. Multilevel regulation of the circadian clock. Nat. Rev. Mol. Cell. Biol. 2000, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.M.; Cheng, Y.; Kapoor, S.; Reilly, D.; Price, T.S.; Fitzgerald, G.A. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc. Natl. Acad. Sci. USA 2007, 104, 3450–3455. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Chrousos, G.P.; Kino, T. Circadian rhythm transcription factor clock regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: Potential physiological implications. FASEB J. 2009, 23, 1572–1583. [Google Scholar] [CrossRef]
- Murayama, Y.; Yahagi, N.; Takeuchi, Y.; Aita, Y.; Mehrazad Saber, Z.; Wada, N.; Li, E.; Piao, X.; Sawada, Y.; Shikama, A.; et al. Glucocorticoid receptor suppresses gene expression of rev-erbalpha (nr1d1) through interaction with the clock complex. FEBS Lett. 2019, 593, 423–432. [Google Scholar] [CrossRef]
- Sainte-Marie, Y.; Nguyen Dinh Cat, A.; Perrier, R.; Mangin, L.; Soukaseum, C.; Peuchmaur, M.; Tronche, F.; Farman, N.; Escoubet, B.; Benitah, J.P.; et al. Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J. 2007, 21, 3133–3141. [Google Scholar] [CrossRef]
- Richardson, R.V.; Rog-Zielinska, E.A.; Thomson, A.J.W.; Moran, C.M.; Kenyon, C.J.; Gray, G.A.; Chapman, K.E. Pathological cardiac remodeling caused by cardiomyocyte/vascular smooth muscle glucocorticoid receptor deficiency. Cardiovasc. Res. 2014, 1031, 362. [Google Scholar]
- Zuo, P.; Stanojevic, D.; Colgan, J.; Han, K.; Levine, M.; Manley, J.L. Activation and repression of transcription by the gap proteins hunchback and kruppel in cultured drosophila cells. Genes Dev. 1991, 5, 254–264. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Fisch, S.; Gray, S.; Heymans, S.; Haldar, S.M.; Wang, B.; Pfister, O.; Cui, L.; Kumar, A.; Lin, Z.; Sen-Banerjee, S.; et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 7074–7079. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimo, D.A.; Anand, P.; Liao, X.; Zhu, H.; Shelkay, S.; Artero-Calderon, P.; Zhang, L.; Kirsh, J.; Moore, D.; Rosca, M.G.; et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J. Biol. Chem. 2014, 289, 5914–5924. [Google Scholar] [CrossRef] [PubMed]
- Lavallee, G.; Andelfinger, G.; Nadeau, M.; Lefebvre, C.; Nemer, G.; Horb, M.E.; Nemer, M. The kruppel-like transcription factor klf13 is a novel regulator of heart development. EMBO J. 2006, 25, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; He, B.; Xu, X.; Cidlowski, J.A. Kruppel-like factor 13 is a major mediator of glucocorticoid receptor signaling in cardiomyocytes and protects these cells from DNA damage and death. J. Biol. Chem. 2016, 291, 19374–19386. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Luo, M.J.; Thieringer, R.; Springer, M.S.; Wright, S.D.; Hermanowski-Vosatka, A.; Plump, A.; Balkovec, J.M.; Cheng, K.; Ding, G.J.; Kawka, D.W.; et al. 11beta-hsd1 inhibition reduces atherosclerosis in mice by altering proinflammatory gene expression in the vasculature. Physiol. Genom. 2013, 45, 47–57. [Google Scholar] [CrossRef]
- Hermanowski-Vosatka, A.; Balkovec, J.M.; Cheng, K.; Chen, H.Y.; Hernandez, M.; Koo, G.C.; Le Grand, C.B.; Li, Z.; Metzger, J.M.; Mundt, S.S.; et al. 11beta-hsd1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J. Exp. Med. 2005, 202, 517–527. [Google Scholar] [CrossRef]
- Deuchar, G.A.; McLean, D.; Hadoke, P.W.F.; Brownstein, D.G.; Webb, D.J.; Mullins, J.J.; Chapman, K.; Seckl, J.R.; Kotelevtsev, Y.V. 11beta-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe-/- mice. Endocrinology 2011, 152, 236–246. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernandez-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis--brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782. [Google Scholar] [CrossRef]
- Yang, N.; Caratti, G.; Ince, L.M.; Poolman, T.M.; Trebble, P.J.; Holt, C.M.; Ray, D.W.; Matthews, L.C. Serum cholesterol selectively regulates glucocorticoid sensitivity through activation of jnk. J. Endocrinol. 2014, 223, 155–166. [Google Scholar] [CrossRef]
- Zhang, T.N.; Yang, N.; Goodwin, J.E.; Mahrer, K.; Li, D.; Xia, J.; Wen, R.; Zhou, H.; Zhang, T.; Song, W.L.; et al. Characterization of circular rna and microrna profiles in septic myocardial depression: A lipopolysaccharide-induced rat septic shock model. Inflammation 2019, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.N.; Li, D.; Xia, J.; Wu, Q.J.; Wen, R.; Yang, N.; Liu, C.F. Non-coding rna: A potential biomarker and therapeutic target for sepsis. Oncotarget 2017, 8, 91765–91778. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shi, X.L.; Zhang, B.L.; Rong, J.; Zhang, T.N.; Xu, W.; Liu, C.F. The trend of beta3-adrenergic receptor in the development of septic myocardial depression: A lipopolysaccharide-induced rat septic shock model. Cardiology 2018, 139, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad Sci. USA 2013, 110, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Zhou, H.; Sessa, W.C. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by dexamethasone after lps. PLoS ONE 2014, 9, e108126. [Google Scholar] [CrossRef] [PubMed]
- Dschietzig, T.; Brecht, A.; Bartsch, C.; Baumann, G.; Stangl, K.; Alexiou, K. Relaxin improves tnf-alpha-induced endothelial dysfunction: The role of glucocorticoid receptor and phosphatidylinositol 3-kinase signalling. Cardiovasc. Res. 2012, 95, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; He, Y.H.; Zhang, G.S.; Luo, M.S.; Huang, Y.; Wu, X.Q.; Liu, S.M.; Luo, J.D.; Chen, M.S. Endogenous glucocorticoids inhibit myocardial inflammation induced by lipopolysaccharide: Involvement of regulation of histone deacetylation. J. Cardiovasc. Pharmacol. 2012, 60, 33–41. [Google Scholar] [CrossRef]
- Abraham, M.N.; Jimenez, D.M.; Fernandes, T.D.; Deutschman, C.S. Cecal ligation and puncture alters glucocorticoid receptor expression. Crit. Care Med. 2018, 46, 797–804. [Google Scholar] [CrossRef]
- Xue, Q.; Dasgupta, C.; Chen, M.; Zhang, L. Foetal hypoxia increases cardiac at(2)r expression and subsequent vulnerability to adult ischaemic injury. Cardiovasc. Res. 2011, 89, 300–308. [Google Scholar] [CrossRef]
- Lv, J.; Ma, Q.; Dasgupta, C.; Xu, Z.; Zhang, L. Antenatal hypoxia and programming of glucocorticoid receptor expression in the adult rat heart. Front Physiol. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.R.; Ma, Q.; Dasgupta, C.; Meng, X.; Zhang, L. Microrna-210 suppresses glucocorticoid receptor expression in response to hypoxia in fetal rat cardiomyocytes. Oncotarget 2017, 8, 80249–80264. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.H.; Han, Q.B.; Dong, G.T.; Yue, R.Q.; Ren, X.C.; Liu, J.X.; Liu, L.; Luo, P.; Zhou, H. Panax ginseng polysaccharide protected h9c2 cardiomyocyte from hypoxia/reoxygenation injury through regulating mitochondrial metabolism and risk pathway. Front Physiol. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Strom, J.; Chen, Q.M. Dexamethasone induces transcriptional activation of bcl-xl gene and inhibits cardiac injury by myocardial ischemia. Eur. J. Pharmacol. 2011, 668, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Patterson, A.J.; Xiao, D.; Zhang, L. Glucocorticoid modulates angiotensin ii receptor expression patterns and protects the heart from ischemia and reperfusion injury. PLoS ONE 2014, 9, e106827. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphism | Glucocorticoid Sensitivity | Involved Risk Factors/Disease | References |
|---|---|---|---|
| ER22/23EK | Decreased | Lower risk of type 2 diabetes mellitus and cardiovascular disease | [50] |
| N363S | Increased | Obesity, type 2 diabetes, coronary artery disease | [54,55,56,57,58] |
| GR-9β | Decreased | Decreased total cholesterol levels and increased HDL cholesterol levels, regulation of human blood pressure | [59] |
| BclI | Increased | Hypertension, adiposity, obesity, atherosclerosis | [60,61,62,63,64,65,66,67] |
| D401H | Increased | Hypertension, diabetes, accumulation of visceral fat | [68] |
| A714Q | Decreased | Hypoglycemia, hypertension | [69] |
| F737L | Decreased | Hypertension, hypokalemia | [70] |
| F774S | Decreased | Hypoglycemia, hypertension | [71] |
| V575G | Decreased | Hypertension, hypokalemia | [72] |
| D641V | Decreased | Hypertension, hypokalemic alkalosis | [73] |
| G679S | Decreased | Hypertension, fatigue | [74] |
| Animal Model | Study Type | GR Knock Out Condition | Outcomes | Reference |
|---|---|---|---|---|
| Mouse | In vivo | Global | Mice died soon after birth because of organ dysfunction. | [76] |
| Mouse | In vivo | Heart-specific | Mice died prematurely from pathological cardiac hypertrophy. | [78] |
| Mouse | In vitro | Heart-specific | Expression of several key genes regarding cardiac contractility, cardiomyocyte survival, and inflammation changed. | [78] |
| Piglet | In vitro | N/A | GR-related pathways that participated in the regulation of myocyte size. | [80] |
| Piglet | In vitro | N/A | GR-related pathways were related with myocyte structural maturation. | [81] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Zhang, T.-N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells 2019, 8, 1227. https://doi.org/10.3390/cells8101227
Liu B, Zhang T-N, Knight JK, Goodwin JE. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells. 2019; 8(10):1227. https://doi.org/10.3390/cells8101227
Chicago/Turabian StyleLiu, Bing, Tie-Ning Zhang, Jessica K. Knight, and Julie E. Goodwin. 2019. "The Glucocorticoid Receptor in Cardiovascular Health and Disease" Cells 8, no. 10: 1227. https://doi.org/10.3390/cells8101227
APA StyleLiu, B., Zhang, T.-N., Knight, J. K., & Goodwin, J. E. (2019). The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells, 8(10), 1227. https://doi.org/10.3390/cells8101227