Late Onset of Estrogen Therapy Impairs Carotid Function of Senescent Females in Association with Altered Prostanoid Balance and Upregulation of the Variant ERα36



 , and
, and
Abstract
1. Introduction
2. Material and Methods
2.1. Animal Models and Hormonal Treatment
2.2. Vascular Function Study
2.3. Quantitative Real-Time PCR (qPCR) for Detection of COX and eNOS Expression
2.4. Measurement of Prostanoids Release from Vascular Segments
2.5. Western Blot Analysis
2.6. Methylation Status of Gene Encoding ERα and Expression of Splicing Variants
2.7. Data Analysis and Statistics
3. Results
3.1. Basic Parameters for Ovariectomy and Estrogen Treatment Efficacy
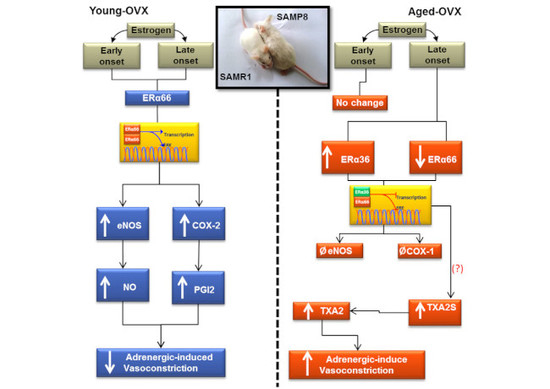
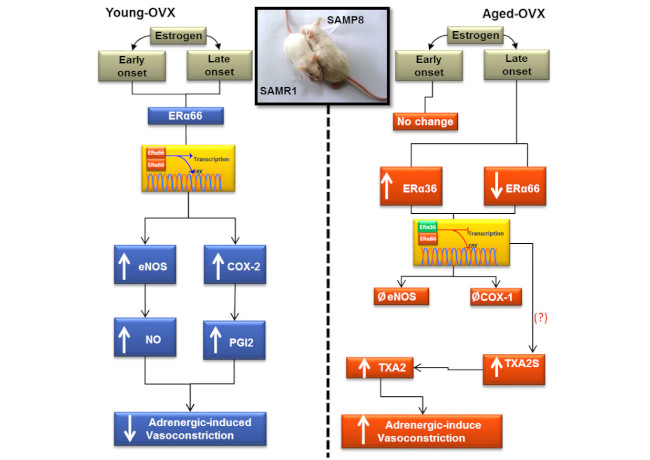
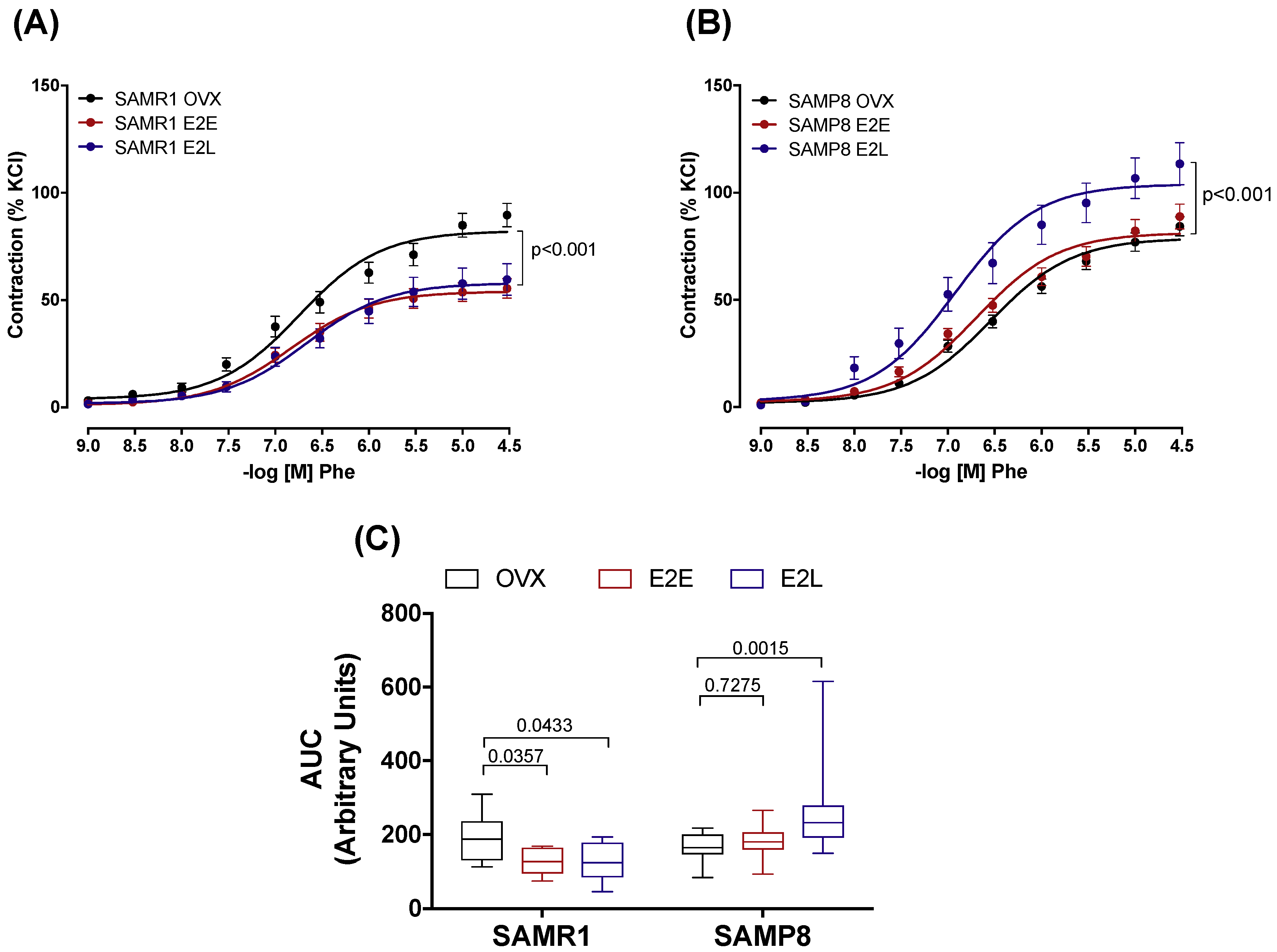
3.2. Early- and Late-Onset Estrogen Treatments Promote Different Vasoconstrictor Effects in Response to Adrenergic Stimulus in SAMR1 and SAMP8 Ovariectomized Female Mice
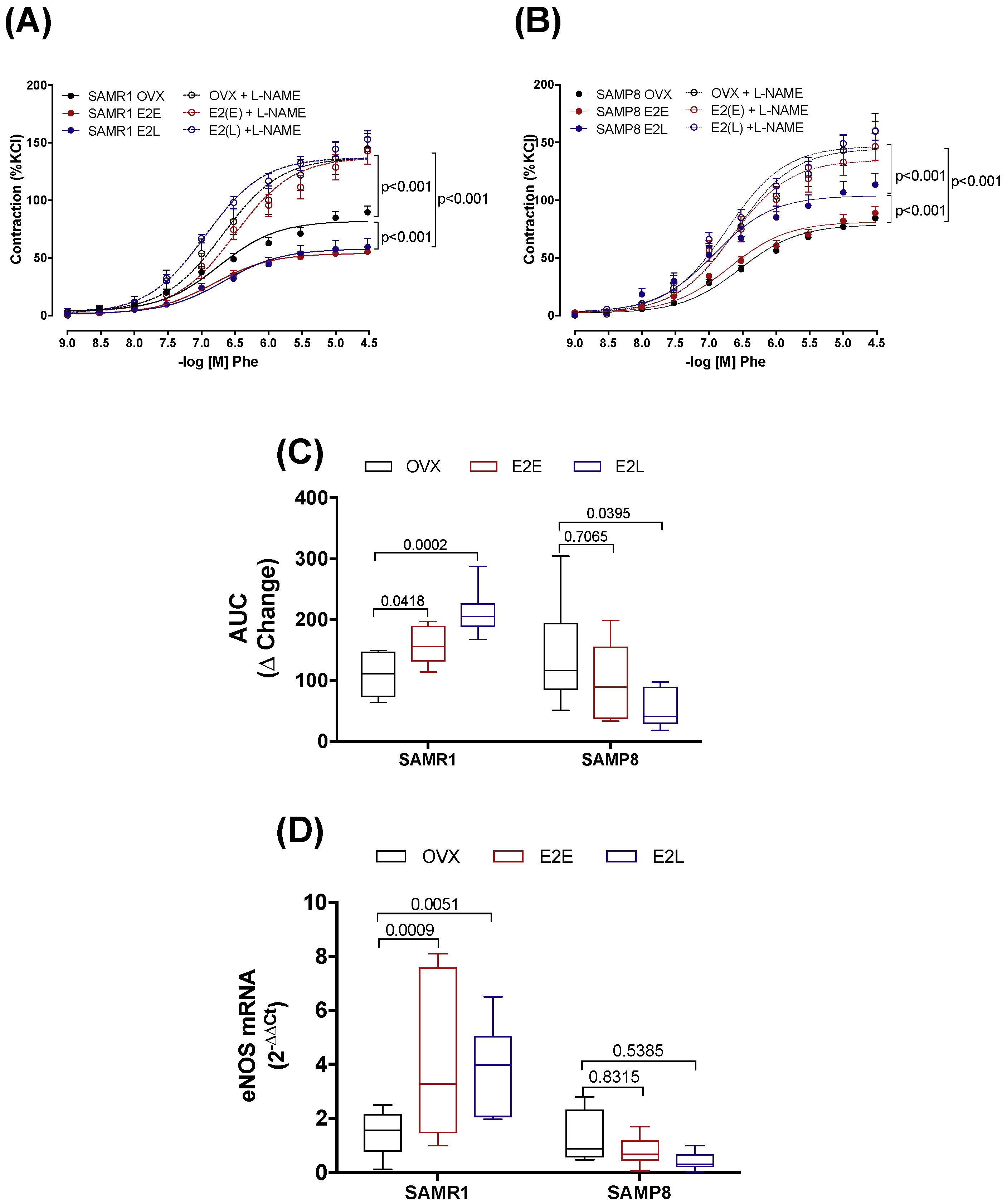
3.3. Estrogen Differently Regulates the Contribution of NO/Superoxide Anion (O2−) Pathways to Phe Contraction in Non-Senescent (SAMR1) and Senescent (SAMP8) Carotids
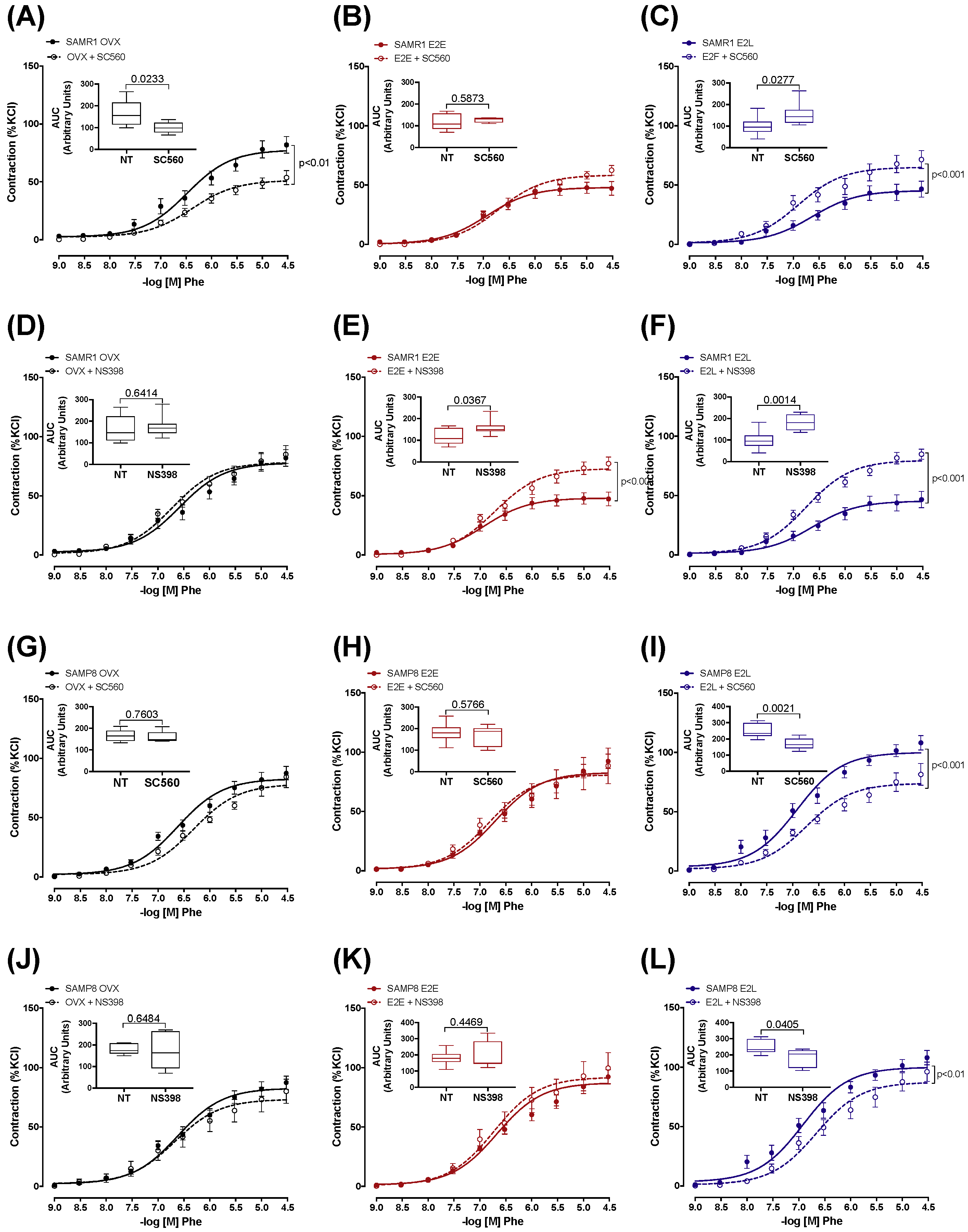
3.4. Senescence and Onset of Estrogen Therapy Play an Important Role in Regulating the Contribution of Vasodilator and Vasoconstrictor Prostanoids to Phe Contraction
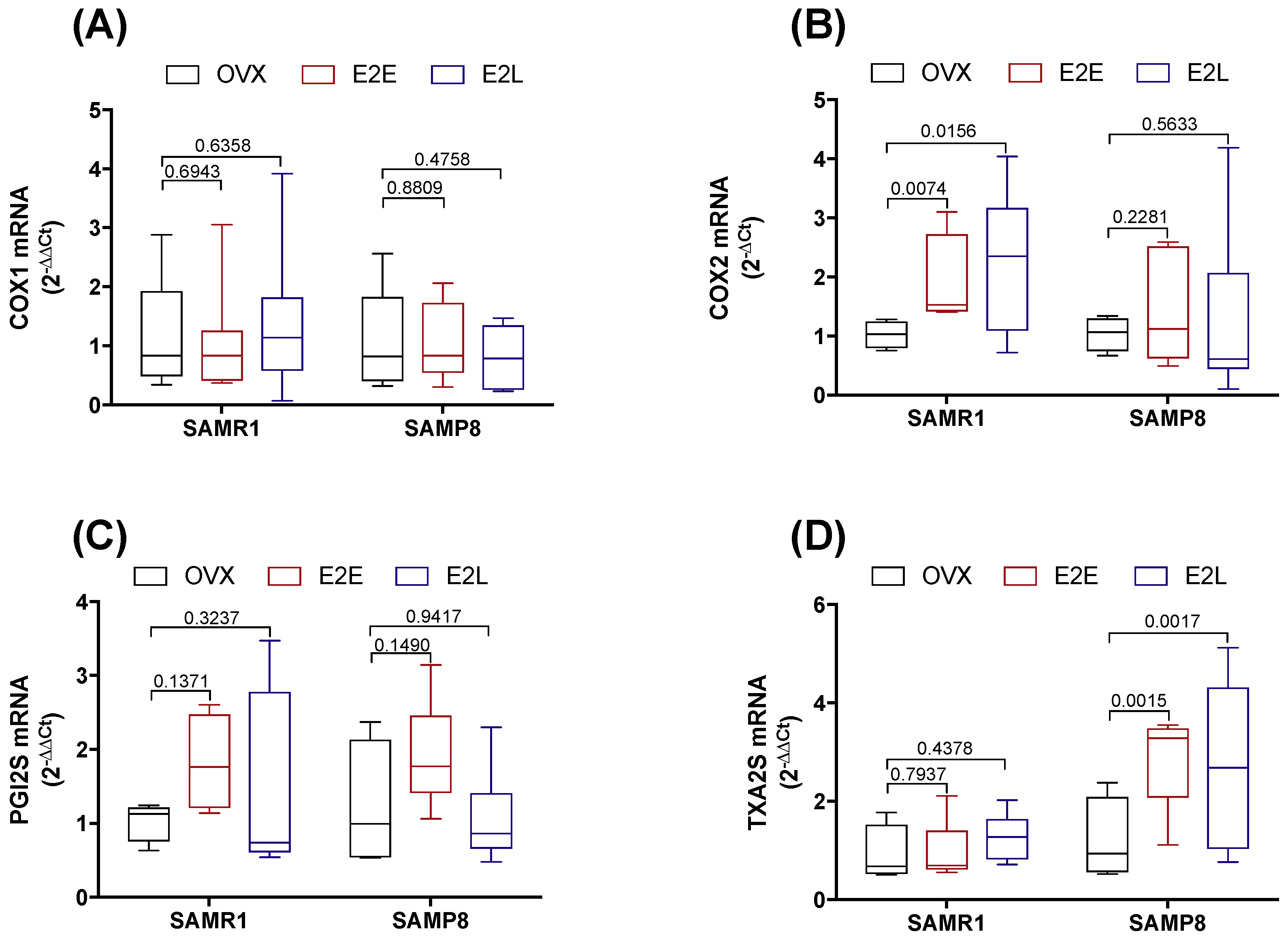
3.5. Dual Effect of COX-1 and COX-2 in Prostanoids Production
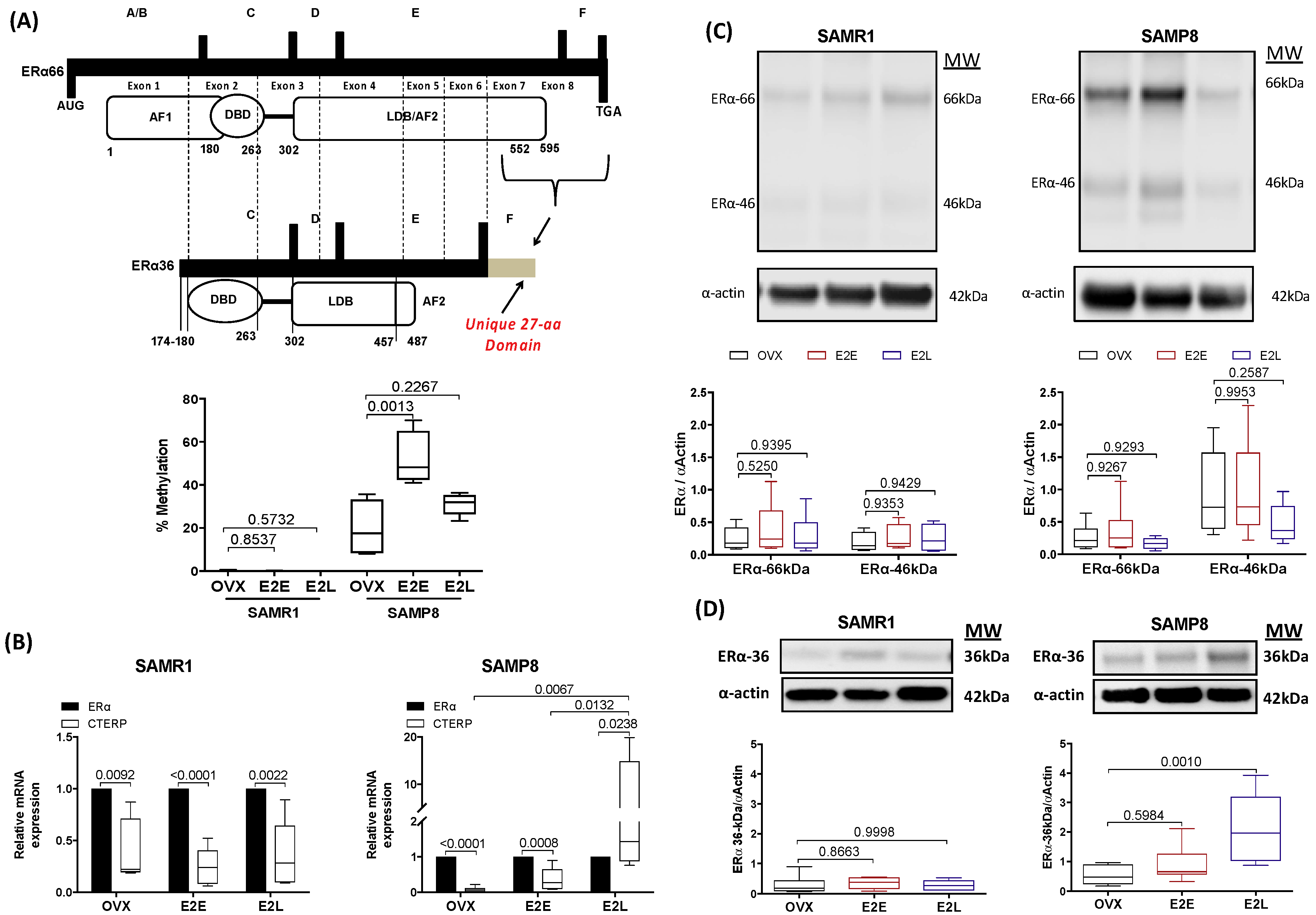
3.6. Late-Onset Estrogen Treatment Increases Alternative Splicing of Estrogen Receptor Alpha (ERα36) in Aging
4. Discussion
5. Translational Perspective
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCA | Common carotid artery |
| OVX | Ovariectomized |
| SAMP8 | Senescence-accelerated mice prone–8 |
| SAMR1 | Senescence-accelerated mice resistant-1 |
| E2 | Estrogen |
| E2E | Early estrogen treatment |
| E2L | Late estrogen treatment |
| Phe | Phenylephrine |
| ACh | Acetylcholine |
| NPS | Sodium nitroprusside |
| ERα | Estrogen receptor alpha |
| COX-1 | Cyclooxygenase 1 |
| COX-2 | Cyclooxygenase 2 |
| PGI2 | Prostacyclin |
| TXA2 | Thromboxane A2 |
References
- Comhaire, F. Hormone replacement therapy, and longevity. Andrologia 2016, 48, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.J.; Willett, W.C.; Colditz, G.A.; Rosner, B.; Speizer, F.E.; Hennekens, C.H. A prospective study of postmenopausal estrogen therapy and coronary heart disease. N. Engl. J. Med. 1985, 313, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Filgueira, F.; Lobato, N.; Ceravolo, G.; Dantas, A.P.; Fortes, Z.; Webb, C.; Tostes, R.; Carvalho, M.H. Characterization of the relaxant response to equilin in rat mesenteric arteries. FASEB J. 2010, 24, 575–577. [Google Scholar]
- Ceravolo, G.S.; Filgueira, F.P.; Costa, T.J.; Lobato, N.S.; Chignalia, A.Z.; Araujo, P.X.; Tostes, R.C.; Dantas, A.P.; Fortes, Z.B.; Carvalho, M.H.C. Conjugated equine estrogen treatment corrected the exacerbated aorta oxidative stress in ovariectomized spontaneously hypertensive rats. Steroids 2013, 78, 341–346. [Google Scholar] [CrossRef]
- Novella, S.; Heras, M.; Hermenegildo, C.; Dantas, A.P. Effects of estrogen on vascular inflammation: A matter of timing. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2035–2042. [Google Scholar] [CrossRef]
- Dantas, A.P.V.; Scivoletto, R.; Fortes, Z.B.; Nigro, D.; Carvalho, M.H.C. Influence of Female Sex Hormones on Endothelium-Derived Vasoconstrictor Prostanoid Generation in Microvessels of Spontaneously Hypertensive Rats. Hypertension 1999, 34, 914–919. [Google Scholar] [CrossRef]
- Dantas, A.P.V.; Tostes, R.C.; Nigro, D.; Fortes, Z.B.; Carvalho, M.H.C. In vivo evidence for the antioxidant potential of estrogen in spontaneously hypertensive rats. Hypertension 2002, 39, 405–411. [Google Scholar] [CrossRef]
- Araujo, P.X.; Costa, T.J.; Echem, C.; de Oliveira, M.; Santos-Eichler, R.A.; Colli, L.G.; Jiménez-Altayó, F.; Vila, E.; Akamine, E.H.; Dantas, A.P.; et al. Treatment with Standard and Low Dose of Conjugated Equine Estrogen Differentially Modulates Estrogen Receptor Expression and Response to Angiotensin II in Mesenteric Venular Bed of Surgically Postmenopausal Hypertensive Rats. J. Pharmacol. Exp. Ther. 2017, 362, 98–107. [Google Scholar] [CrossRef]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [PubMed]
- Grodstein, F.; Manson, J.E.; Stampfer, M.J. Hormone therapy and coronary heart disease: The role of time since menopause and age at hormone initiation. J. Womens Heal. 2006, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Harman, S.M.; Vittinghoff, E.; Brinton, E.A.; Budoff, M.J.; Cedars, M.I.; Lobo, R.A.; Merriam, G.R.; Miller, V.M.; Naftolin, F.; Pal, L.; et al. Timing and duration of menopausal hormone treatment may affect cardiovascular outcomes. Am. J. Med. 2011, 124, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dustin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Izzo, C.; Carrizzo, A.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Calabrese, M.; De Simone, E.; Sciarretta, S.; Frati, G.; Oliveti, M.; et al. The impact of aging on cardio and cerebrovascular diseases. Int. J. Mol. Sci. 2018, 19, 481. [Google Scholar] [CrossRef] [PubMed]
- Novensa, L.; Novella, S.; Medina, P.; Segarra, G.; Castillo, N.; Heras, M.; Hermenegildo, C.; Dantas, A.P. Aging Negatively Affects Estrogens-Mediated Effects on Nitric Oxide Bioavailability by Shifting ER alpha/ER beta Balance in Female Mice. PLoS ONE 2011, 6, e25335. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef]
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensà, L.; Bueno, C.; Heras, M.; Hermenegildo, C.; Medina, P. Gathering of aging and estrogen withdrawal in vascular dysfunction of senescent accelerated mice. Exp. Gerontol. 2010, 45, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Onetti, Y.; Jiménez-Altayó, F.; Heras, M.; Vila, E.; Dantas, A.P. Western-type diet induces senescence, modifies vascular function in non-senescence mice and triggers adaptive mechanisms in senescent ones. Exp. Gerontol. 2013, 48, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.A.; Johnson, K.M. Menopause. Med. Clin. N. Am. 2015, 99, 521–534. [Google Scholar] [CrossRef]
- Xing, D.; Nozell, S.; Chen, Y.F.; Hage, F.; Oparil, S. Estrogen and mechanisms of vascular protection. Arter. Thromb. Vasc. Biol. 2009, 29, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Lee, J.S.; Pechenino, A.S.; Knowlton, A.A. Estrogen, aging and the cardiovascular system. Futur. Cardiol. 2009, 5, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.F. Effects of central arterial aging on the structure and function of the peripheral vasculature: Implications for end-organ damage. J. Appl. Physiol. 2008, 105, 1652–1660. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef]
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensa, L.; Heras, M.; Hermenegildo, C.; Medina, P. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age (Omaha) 2013, 35, 117–128. [Google Scholar] [CrossRef]
- Vidal-Gómez, X.; Novella, S.; Pérez-Monzó, I.; Garabito, M.; Dantas, A.P.; Segarra, G.; Hermenegildo, C.; Medina, P. Decreased bioavailability of nitric oxide in aorta from ovariectomized senescent mice: Role of cyclooxygenase. Exp. Gerontol. 2016, 76, 1–8. [Google Scholar] [CrossRef]
- Virdis, A.; Ghiadoni, L.; Pinto, S.; Lombardo, M.; Petraglia, F.; Gennazzani, A.; Buralli, S.; Taddei, S.; Salvetti, A. Mechanisms responsible for endothelial dysfunction associated with acute estrogen deprivation in normotensive women. Circulation 2000, 101, 2258–2263. [Google Scholar] [CrossRef] [PubMed]
- Rubanyi, G.M.; Freay, A.D.; Kauser, K.; Sukovich, D.; Burton, G.; Lubahn, D.B.; Couse, J.F.; Curtis, S.W.; Korach, K.S. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J. Clin. Investig. 1997, 99, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Rubanyi, G.M.; Johns, A.; Kauser, K. Effect of estrogen on endothelial function and angiogenesis. Vasc. Pharmacol. 2002, 38, 89–98. [Google Scholar] [CrossRef]
- Costa, T.J.; Ceravolo, G.S.; Dos Santos, R.A.; De Oliveira, M.A.; Araújo, P.X.; Giaquinto, L.R.; Tostes, R.C.; Akamine, E.H.; Fortes, Z.B.; Dantas, A.P.; et al. Association of testosterone with estrogen abolishes the beneficial effects of estrogen treatment by increasing ROS generation in aorta endothelial cells. Am. J. Physiol. - Hear. Circ. Physiol. 2015, 308, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, A.; Bower, J.K.; McFetridge-Durdle, J.; Blumenthal, J.A.; Newby, L.K.; Hinderliter, A.L. Age moderates the short-term effects of transdermal 17beta-estradiol on endothelium-dependent vascular function in postmenopausal women. Arter. Thromb. Vasc. Biol. 2007, 27, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Montoya, N.; Giraldo, J.; Vila, E. Ageing affects nitric oxide synthase, cyclooxygenase and oxidative stress enzymes expression differently in mesenteric resistance arteries. Aut. Autacoid Pharmacol. 2005, 25, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Garabito, M.; Jiménez-Altayó, F.; Onetti, Y.; Sabate, M.; Vila, E.; Dantas, A.P. Sex differences in angiotensin II responses contribute to a differential regulation of cox-mediated vascular dysfunction during aging. Exp. Gerontol. 2016, 85, 71–80. [Google Scholar] [CrossRef]
- Hermenegildo, C.; Oviedo, P.J.; Cano, A. Cyclooxygenases regulation by estradiol on endothelium. Curr. Pharm. Des. 2006, 12, 205–215. [Google Scholar] [CrossRef]
- Mueck, A.O.; Seeger, H.; Lüdtke, R.; Gräser, T.; Wallwiener, D. Effect on biochemical vasoactive markers during postmenopausal hormone replacement therapy: Estradiol versus estradiol/dienogest. Maturitas 2001, 38, 305–313. [Google Scholar] [CrossRef]
- Egan, K.M.; Lawson, J.A.; Fries, S.; Koller, B.; Rader, D.J.; Smyth, E.M.; Fitzgerald, G.A. COX-2-derived prostacyclin confers atheroprotection on female mice. Science (80-.) 2004, 306, 1954–1957. [Google Scholar] [CrossRef]
- Akarasereenont, P.; Techatraisak, K.; Thaworn, A.; Chotewuttakorn, S. The induction of cyclooxygenase-2 by 17beta-estradiol in endothelial cells is mediated through protein kinase C. Inflamm. Res. 2000, 49, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.L.; Aldrighi, J.M.; Gebara, O.E.; Rocha, T.R.; D’Amico, E.; Rosano, G.M.; Ramires, J.A. Postmenopausal hormone replacement therapy increases plasmatic thromboxane beta 2. Int. J. Cardiol. 2005, 99, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, A.; Mata, M.; Laguna-Fernandez, A.; Novella, S.; Oviedo, P.J.; García-Pérez, M.A.; Tarín, J.J.; Cano, A.; Hermenegildo, C. Estradiol stimulates vasodilatory and metabolic pathways in cultured human endothelial cells. PLoS ONE 2009, 4, e8242. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.; Krust, A.; Karas, R.H.; Dupont, S.; Aronovitz, M.; Chambon, P.; Mendelsohn, M.E. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ. Res. 2002, 90, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Billon-Galés, A.; Fontaine, C.; Douin-Echinard, V.; Delpy, L.; Berges, H.; Calippe, B.; Lenfant, F.; Laurell, H.; Guéry, J.C.; Gourdy, P.; et al. Endothelial estrogen receptor-alpha plays a crucial role in the atheroprotective action of 17beta-estradiol in low-density lipoprotein receptor-deficient mice. Circulation 2009, 120, 2567–2576. [Google Scholar] [CrossRef]
- Sumi, D.; Ignarro, L.J. Estrogen-related receptor alpha 1 upregulates endothelial nitric oxide synthase expression. Proc. Natl. Acad. Sci. USA 2003, 100, 14451–14456. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Raz, L.; Wang, R.; Han, D.; De Sevilla, L.; Yang, F.; Vadlamudi, R.K.; Brann, D.W. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J. Neurosci. 2009, 29, 13823–13836. [Google Scholar] [CrossRef]
- Sobrino, A.; Oviedo, P.J.; Novella, S.; Laguna-Fernandez, A.; Bueno, C.; García-Pérez, M.A.; Tarín, J.J.; Cano, A.; Hermenegildo, C. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor-{alpha}. J. Mol. Endocrinol. 2010, 44, 237–246. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Patkar, S.; Farr, T.D.; Cooper, E.; Dowell, F.J.; Carswell, H. V Differential vasoactive effects of oestrogen, oestrogen receptor agonists and selective oestrogen receptor modulators in rat middle cerebral artery. Neurosci. Res. 2011, 71, 78–84. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAMR1 | SAMP8 | |||||
|---|---|---|---|---|---|---|
| OVX | E2E | E2L | OVX | E2E | E2L | |
| Uterus wet weight (mg/cm) | 30.1 ± 4.5 | 49.5 ± 5.4 * | 56.1 ± 7.1 * | 14.0 ± 1.7 | 42.4 ± 5.7 * | 40.3 ± 5.5 * |
| Uterus dry weight (mg/cm) | 7.9 ± 1.1 | 12.3 ± 1.0 * | 13.9 ± 1.6 * | 4.3 ± 0.8 | 12.1 ± 1.8 * | 11.0 ± 1.2 * |
| 17β-estradiol (pg/mL) | 2.1 ± 0.5 | 4.2 ± 0.9 * | 7.0 ± 1.2 * | 1.2 ± 0.3 | 9.8 ± 1.4 * | 7.4 ± 0.9 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Januário Costa, T.; Jiménez-Altayó, F.; Echem, C.; Akamine, E.H.; Tostes, R.; Vila, E.; Dantas, A.P.; Catelli de Carvalho, M.H. Late Onset of Estrogen Therapy Impairs Carotid Function of Senescent Females in Association with Altered Prostanoid Balance and Upregulation of the Variant ERα36. Cells 2019, 8, 1217. https://doi.org/10.3390/cells8101217
Januário Costa T, Jiménez-Altayó F, Echem C, Akamine EH, Tostes R, Vila E, Dantas AP, Catelli de Carvalho MH. Late Onset of Estrogen Therapy Impairs Carotid Function of Senescent Females in Association with Altered Prostanoid Balance and Upregulation of the Variant ERα36. Cells. 2019; 8(10):1217. https://doi.org/10.3390/cells8101217
Chicago/Turabian StyleJanuário Costa, Tiago, Francesc Jiménez-Altayó, Cinthya Echem, Eliana Hiromi Akamine, Rita Tostes, Elisabet Vila, Ana Paula Dantas, and Maria Helena Catelli de Carvalho. 2019. "Late Onset of Estrogen Therapy Impairs Carotid Function of Senescent Females in Association with Altered Prostanoid Balance and Upregulation of the Variant ERα36" Cells 8, no. 10: 1217. https://doi.org/10.3390/cells8101217
APA StyleJanuário Costa, T., Jiménez-Altayó, F., Echem, C., Akamine, E. H., Tostes, R., Vila, E., Dantas, A. P., & Catelli de Carvalho, M. H. (2019). Late Onset of Estrogen Therapy Impairs Carotid Function of Senescent Females in Association with Altered Prostanoid Balance and Upregulation of the Variant ERα36. Cells, 8(10), 1217. https://doi.org/10.3390/cells8101217