Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. HL-1 Cardiomyocyte Cell Culture and Tachypacing
2.2. Transfection and Drug Treatments
2.3. Calcium Transient Measurements
2.4. Measurements of Mmitochondrial Dysfunction
2.4.1. ATP Measurements
2.4.2. Reactive Oxygen Species (ROS) Measurements
2.4.3. Mitochondrial Morphology Analysis
2.4.4. Mitochondrial Membrane Potential Analysis
2.4.5. Measurement of Mitochondrial Oxygen Consumption Rate
2.5. Protein Extraction and Western Blot Analysis
2.6. Quantitative RT-PCR
2.7. Drosophila Melanogaster Heart Rate Measurements
2.8. Patient Material
2.9. Statistical Analysis
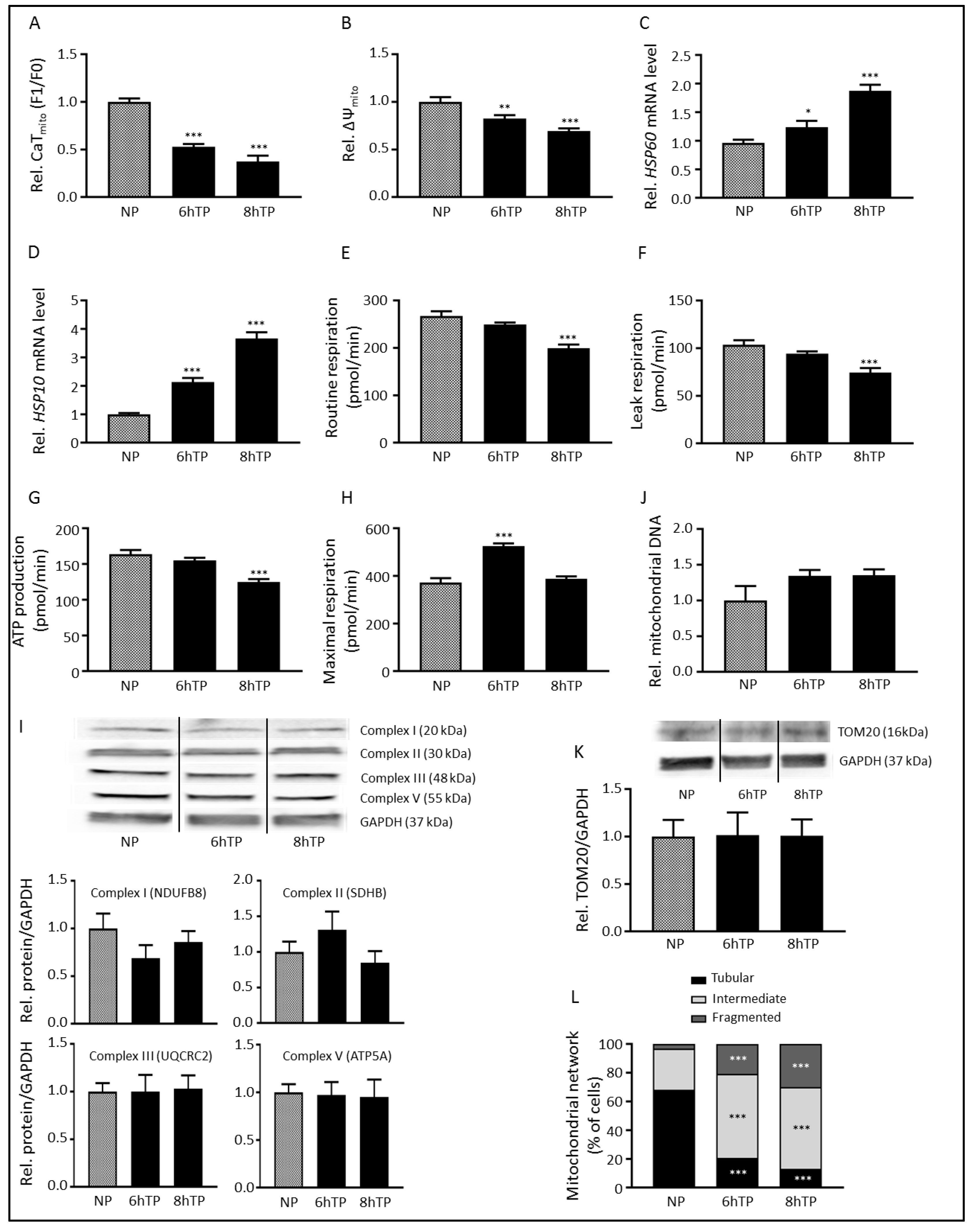
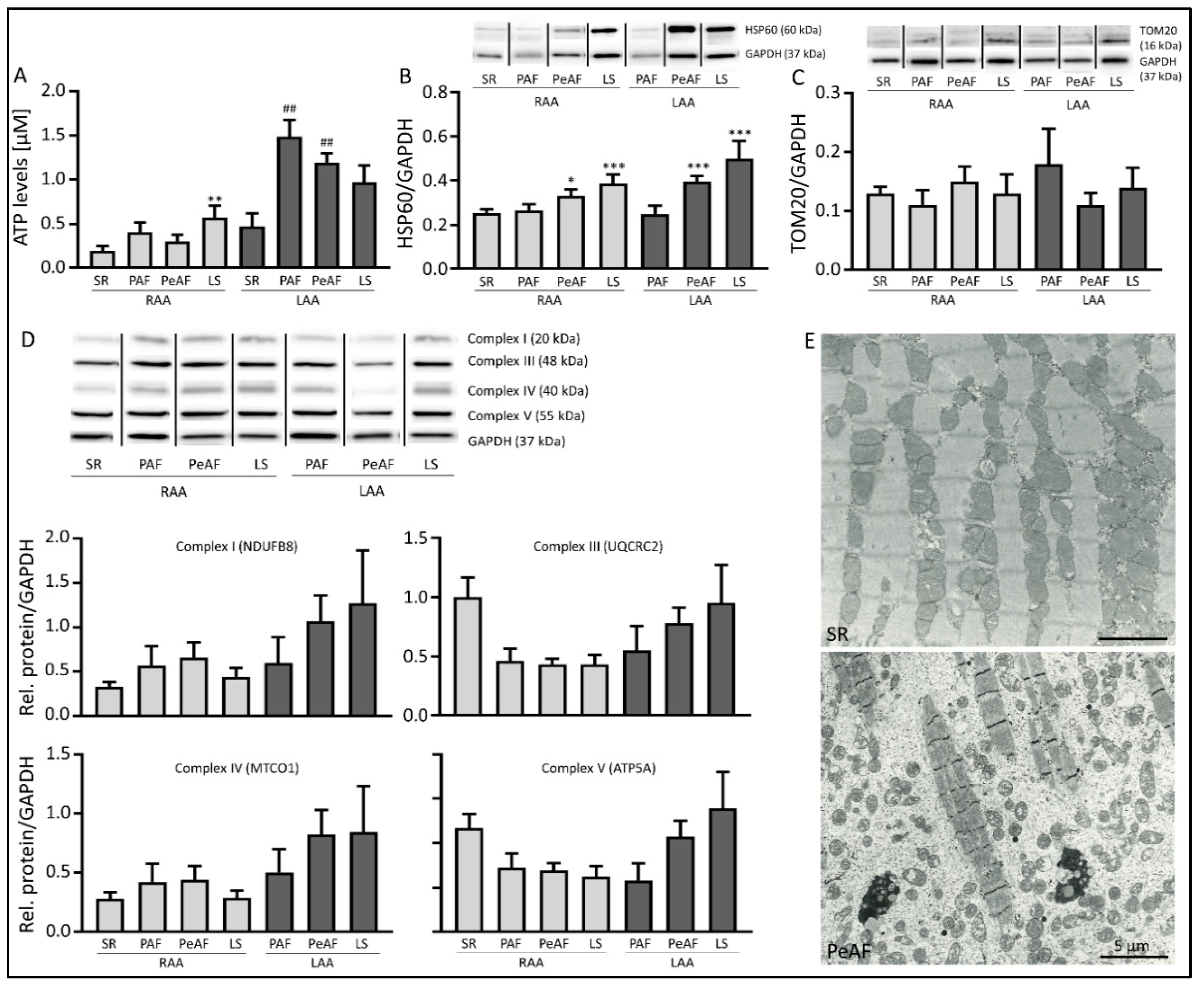
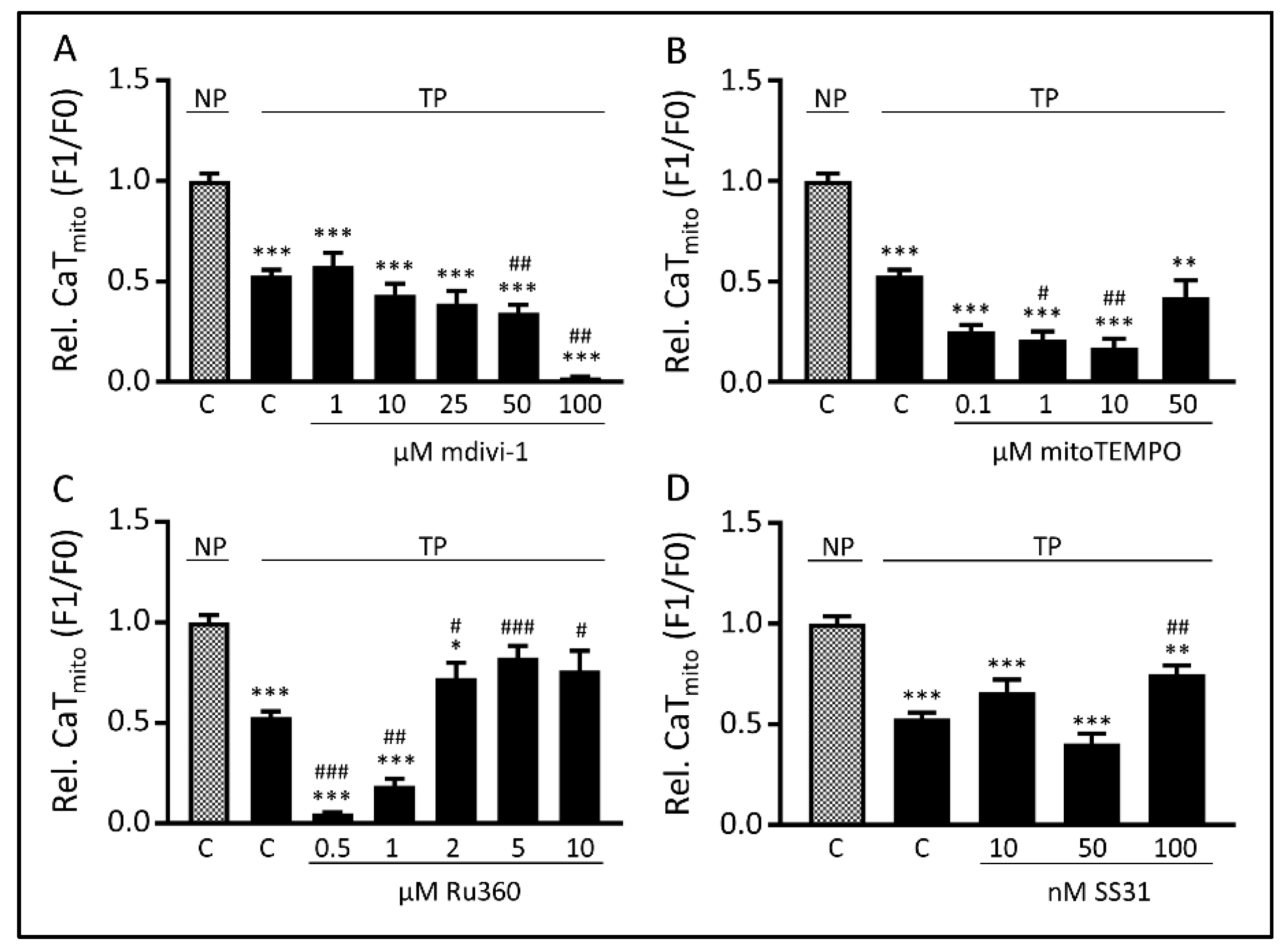
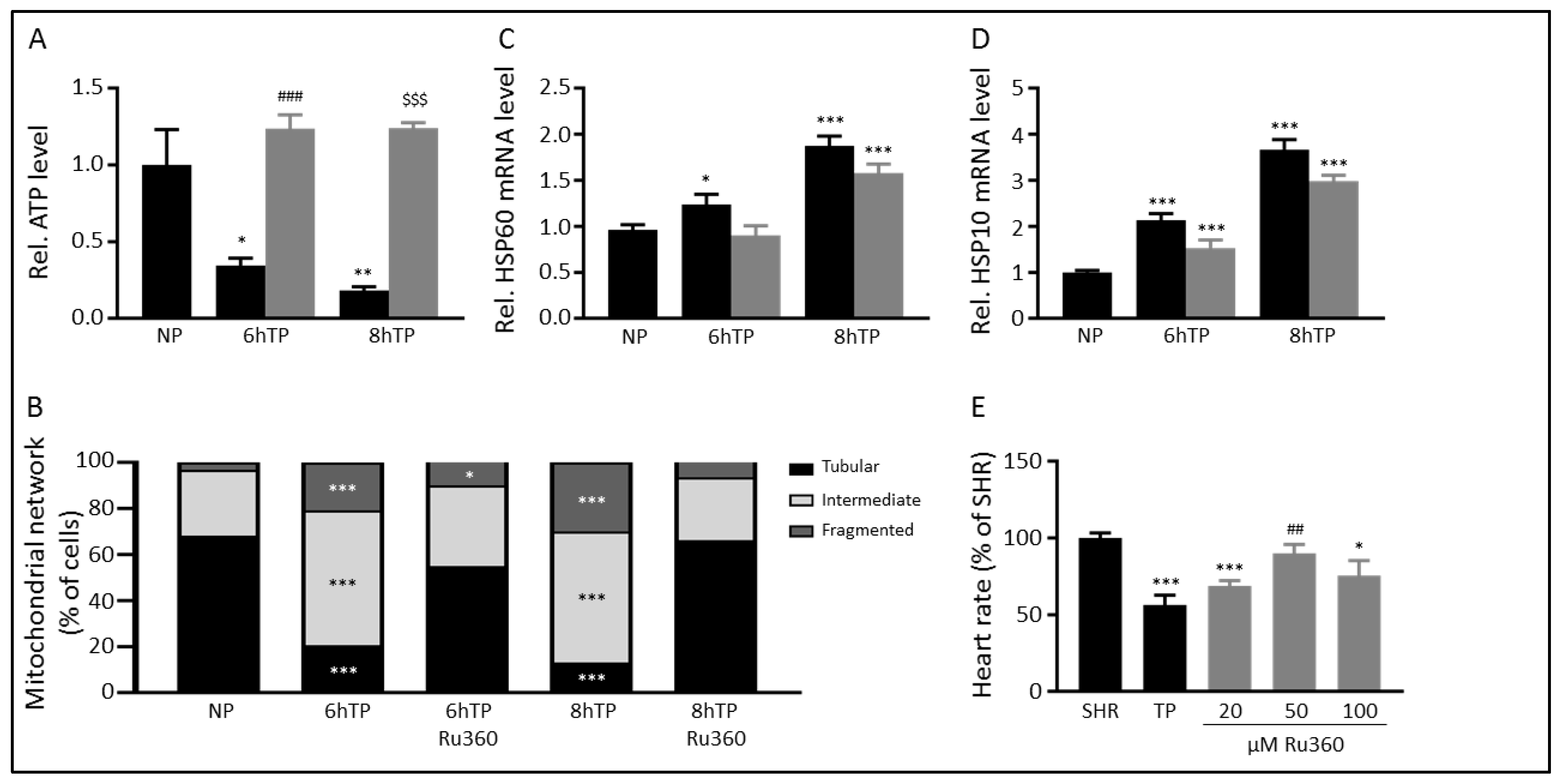
3. Results
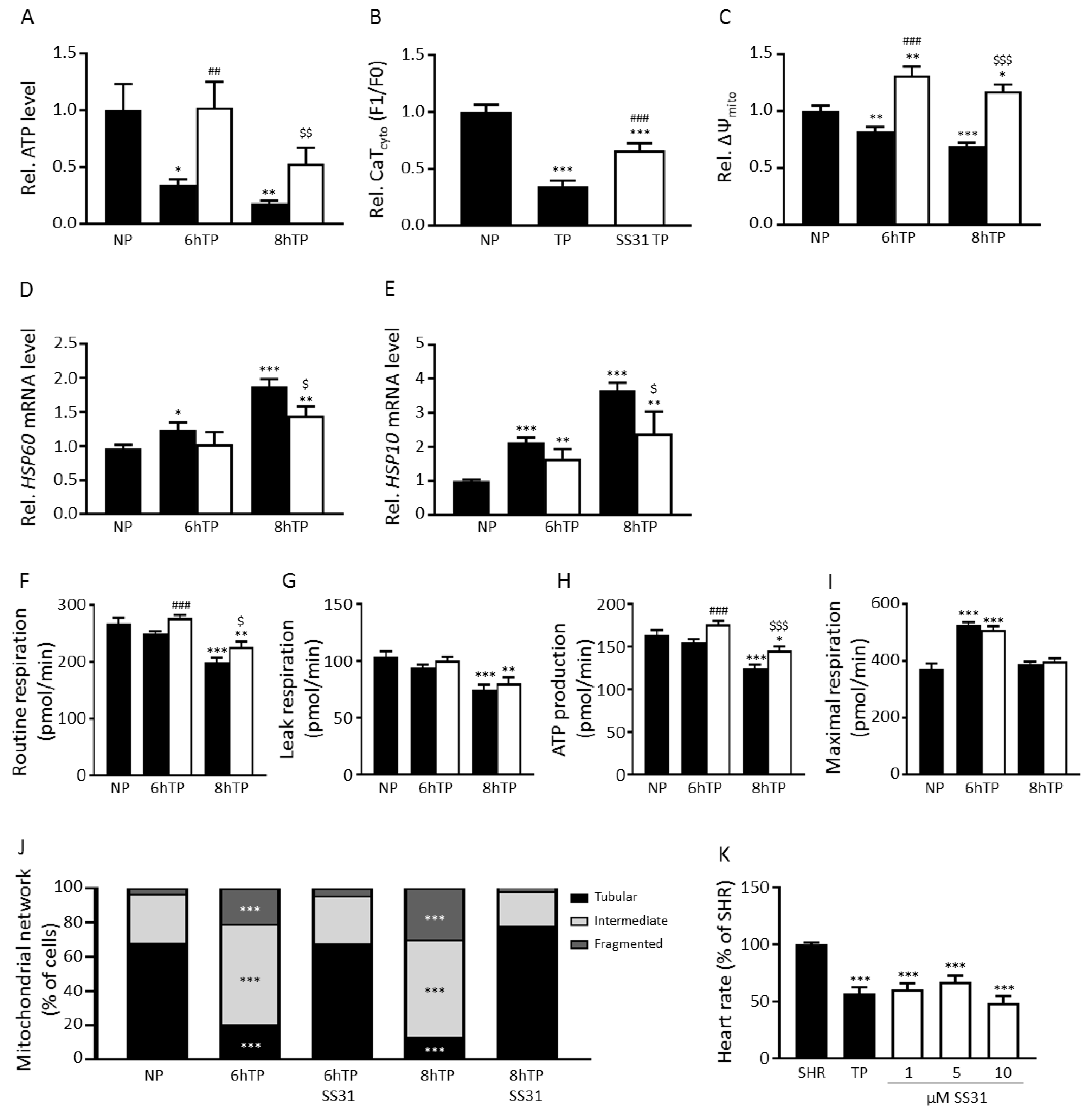
3.1. Tachypacing Induces Mitochondrial Dysfunction and Stress
3.2. Markers of Mitochondrial Dysfunction are Present in AF Patients
3.3. Tachypacing-Induced CaTmito Loss Was Prevented by the Mitochondrial Ca2+ Uniporter Inhibitor Ru360 and Peptide SS31
3.4. Ru360 Protects against Tachypacing-Induced Mitochondrial Dysfunction
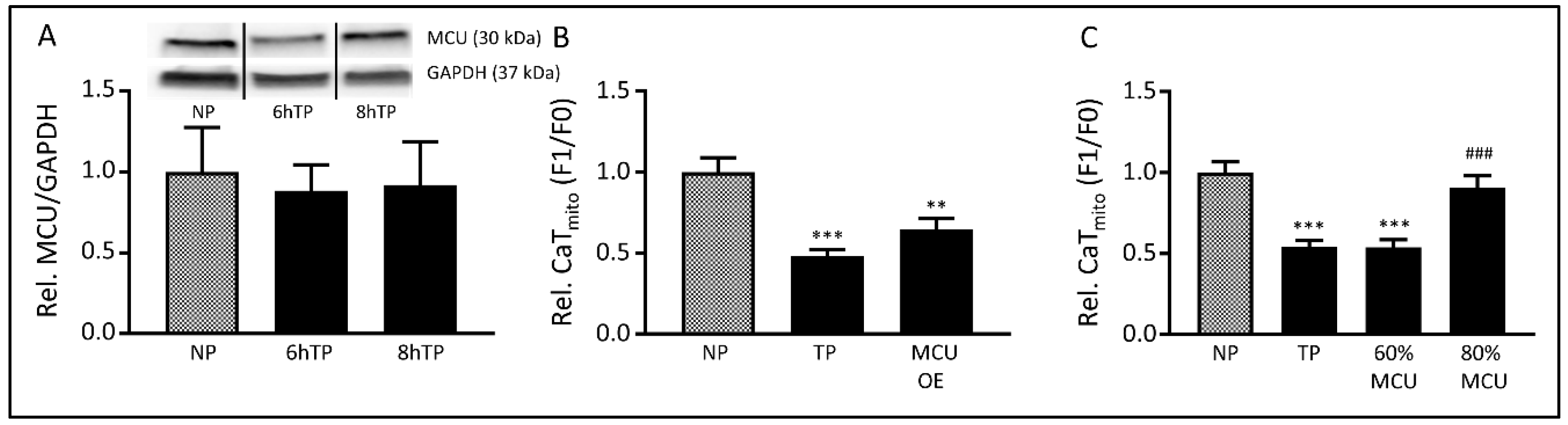
3.5. The MCU may Mediate Tachypacing-Induced Mitochondrial Changes
3.6. SS31 Protects against Tachypacing-Induced Mitochondrial Dysfunction and Stress
4. Discussion
4.1. Mitochondrial Dysfunction and Its Implication in Cardiac Disease
4.2. MCU-Mediated Mitochondrial Dysfunction and Stress in AF
4.3. Amelioration of Tachypacing-Induced Mitochondrial Dysfunction by SS31
4.4. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dobrev, D.; Carlsson, L.; Nattel, S. Novel molecular targets for atrial fibrillation therapy. Nat. Rev. Drug Discov. 2012, 11, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Dobrev, D. Irregular rhythm and atrial metabolism are key for the evolution of proarrhythmic atrial remodeling in atrial fibrillation. Basic Res. Cardiol. 2015, 110, 41. [Google Scholar] [CrossRef] [PubMed]
- Zoni-Berisso, M.; Lercari, F.; Carazza, T.; Domenicucci, S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014, 6, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Buch, E.; Share, M.; Tung, R.; Benharash, P.; Sharma, P.; Koneru, J.; Mandapati, R.; Ellenbogen, K.A.; Shivkumar, K. Long-term clinical outcomes of focal impulse and rotor modulation for treatment of atrial fibrillation: A multicenter experience. Heart Rhythm. 2016, 13, 636–641. [Google Scholar] [CrossRef]
- Winkle, R.A.; Jarman, J.W.; Mead, R.H.; Engel, G.; Kong, M.H.; Fleming, W.; Patrawala, R.A. Predicting atrial fibrillation ablation outcome: The CAAP-AF score. Heart Rhythm. 2016, 13, 2119–2125. [Google Scholar] [CrossRef]
- Jaakkola, S.; Lip, G.Y.; Biancari, F.; Nuotio, I.; Hartikainen, J.E.; Ylitalo, A.; Airaksinen, K.E. Predicting Unsuccessful Electrical Cardioversion for Acute Atrial Fibrillation (from the AF-CVS Score). Am. J. Cardiol. 2017, 119, 749–752. [Google Scholar] [CrossRef]
- Brooks, S.; Metzner, A.; Wohlmuth, P.; Lin, T.; Wissner, E.; Tilz, R.; Rillig, A.; Mathew, S.; Saguner, A.; Heeger, C.; et al. Insights into ablation of persistent atrial fibrillation: Lessons from 6-year clinical outcomes. J. Cardiovasc. Electrophysiol. 2018, 29, 257–263. [Google Scholar] [CrossRef]
- de Groot, N.M.; Houben, R.P.; Smeets, J.L.; Boersma, E.; Schotten, U.; Schalij, M.J.; Crijns, H.; Allessie, M.A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: Epicardial breakthrough. Circulation 2010, 122, 1674–1682. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Mericskay, M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 2016, 109, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Greiser, M.; Lederer, W.J.; Schotten, U. Alterations of atrial Ca(2+) handling as cause and consequence of atrial fibrillation. Cardiovasc. Res. 2011, 89, 722–733. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Babcock, D.F.; Herrington, J.; Goodwin, P.C.; Park, Y.B.; Hille, B. Mitochondrial participation in the intracellular Ca2+ network. J. Cell. Biol. 1997, 136, 833–844. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Camacho, P.; Lechleiter, J.D.; Herman, B. Measurement of intracellular calcium. Physiol. Rev. 1999, 79, 1089–1125. [Google Scholar] [CrossRef]
- Chida, J.; Yamane, K.; Takei, T.; Kido, H. An efficient extraction method for quantitation of adenosine triphosphate in mammalian tissues and cells. Anal. Chim. Acta 2012, 727, 8–12. [Google Scholar] [CrossRef]
- Huang, H.; Frohman, M.A. Visualizing mitochondrial lipids and fusion events in Mammalian cells. Methods Cell Biol. 2012, 108, 131–145. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Lanters, E.A.; van Marion, D.M.; Kik, C.; Steen, H.; Bogers, A.J.; Allessie, M.A.; Brundel, B.J.; de Groot, N.M. HALT & REVERSE: Hsf1 activators lower cardiomyocyt damage; towards a novel approach to REVERSE atrial fibrillation. J. Transl. Med. 2015, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-Te Bulte, L.; Wiersma, M.; Malik, N.U.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD(+) depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1307. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Brundel, B.J.; Ausma, J.; van Gelder, I.C.; Van der Want, J.J.; van Gilst, W.H.; Crijns, H.J.; Henning, R.H. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 2002, 54, 380–389. [Google Scholar] [CrossRef]
- Tanaka, A.; Youle, R.J. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol. Cell 2008, 29, 409–410. [Google Scholar] [CrossRef]
- Liang, H.L.; Sedlic, F.; Bosnjak, Z.; Nilakantan, V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic. Biol. Med. 2010, 49, 1550–1560. [Google Scholar] [CrossRef]
- Ying, W.L.; Emerson, J.; Clarke, M.J.; Sanadi, D.R. Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry 1991, 30, 4949–4952. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Zhang, D.; Ke, L.; Mackovicova, K.; Van Der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef]
- Guzman Mentesana, G.; Baez, A.L.; Lo Presti, M.S.; Dominguez, R.; Cordoba, R.; Bazan, C.; Strauss, M.; Fretes, R.; Rivarola, H.W.; Paglini-Oliva, P. Functional and structural alterations of cardiac and skeletal muscle mitochondria in heart failure patients. Arch. Med. Res. 2014, 45, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur. Heart J. 2014, 35, 3258–3266. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Allessie, M. The “second factor”: A first step toward diagnosing the substrate of atrial fibrillation? J. Am. Coll. Cardiol. 2009, 53, 1192–1193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stiles, M.K.; John, B.; Wong, C.X.; Kuklik, P.; Brooks, A.G.; Lau, D.H.; Dimitri, H.; Roberts-Thomson, K.C.; Wilson, L.; De Sciscio, P.; et al. Paroxysmal lone atrial fibrillation is associated with an abnormal atrial substrate: Characterizing the "second factor". J. Am. Coll. Cardiol. 2009, 53, 1182–1191. [Google Scholar] [CrossRef]
- Swartz, M.F.; Fink, G.W.; Lutz, C.J.; Taffet, S.M.; Berenfeld, O.; Vikstrom, K.L.; Kasprowicz, K.; Bhatta, L.; Puskas, F.; Kalifa, J.; et al. Left versus right atrial difference in dominant frequency, K(+) channel transcripts, and fibrosis in patients developing atrial fibrillation after cardiac surgery. Heart Rhythm. 2009, 6, 1415–1422. [Google Scholar] [CrossRef]
- Caballero, R.; de la Fuente, M.G.; Gomez, R.; Barana, A.; Amoros, I.; Dolz-Gaiton, P.; Osuna, L.; Almendral, J.; Atienza, F.; Fernandez-Aviles, F.; et al. In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J. Am. Coll. Cardiol. 2010, 55, 2346–2354. [Google Scholar] [CrossRef]
- Schaper, J.; Meiser, E.; Stammler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280, 561–573. [Google Scholar] [CrossRef]
- Foskett, J.K.; Philipson, B. The mitochondrial Ca(2+) uniporter complex. J. Mol. Cell. Cardiol. 2015, 78, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Reda, S.; Wolny, M.; Hoppe, U.C. UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia. Pharmaceuticals 2015, 8, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, S.; Wang, S.; Yu, N.; Liu, J. The effect of mitochondrial calcium uniporter on mitochondrial fission in hippocampus cells ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2015, 461, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Crompton, M. Mitochondrial calcium transport. FEBS Lett. 1980, 111, 261–268. [Google Scholar] [CrossRef]
- Quan, X.; Nguyen, T.T.; Choi, S.K.; Xu, S.; Das, R.; Cha, S.K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell. Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef]
- Deheshi, S.; Dabiri, B.; Fan, S.; Tsang, M.; Rintoul, G.L. Changes in mitochondrial morphology induced by calcium or rotenone in primary astrocytes occur predominantly through ros-mediated remodeling. J. Neurochem. 2015, 133, 684–699. [Google Scholar] [CrossRef]
- Zhou, L.; Solhjoo, S.; Millare, B.; Plank, G.; Abraham, M.R.; Cortassa, S.; Trayanova, N.; O’Rourke, B. Effects of regional mitochondrial depolarization on electrical propagation: Implications for arrhythmogenesis. Circ. Arrhythm Electrophysiol. 2014, 7, 143–151. [Google Scholar] [CrossRef]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Schiller, P.W. Novel therapies targeting inner mitochondrial membrane—From discovery to clinical development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, H.; Hao, S.; Chen, J.; Wu, J.; Song, C.; Zhang, M.; Qiao, T.; Li, K. Peptide SS-31 upregulates frataxin expression and improves the quality of mitochondria: Implications in the treatment of Friedreich ataxia. Sci. Rep. 2017, 7, 9840. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Won, K.; Wu, D.; Soong, Y.; Liu, S.; Szeto, H.H.; Hong, M.K. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron. Artery. Dis. 2007, 18, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef]
- Constantinou, C.; Apidianakis, Y.; Psychogios, N.; Righi, V.; Mindrinos, M.N.; Khan, N.; Swartz, H.M.; Szeto, H.H.; Tompkins, R.G.; Rahme, L.G.; et al. In vivo high-resolution magic angle spinning magnetic and electron paramagnetic resonance spectroscopic analysis of mitochondria-targeted peptide in Drosophila melanogaster with trauma-induced thoracic injury. Int. J. Mol. Med. 2016, 37, 299–308. [Google Scholar] [CrossRef]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kaneki, M.; Andreas, J.; Tompkins, R.G.; Martyn, J.A. Novel mitochondria-targeted antioxidant peptide ameliorates burn-induced apoptosis and endoplasmic reticulum stress in the skeletal muscle of mice. Shock 2011, 36, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Meijering, R.A.; Wiersma, M.; van Marion, D.M.; Zhang, D.; Hoogstra-Berends, F.; Dijkhuis, A.J.; Schmidt, M.; Wieland, T.; Kampinga, H.H.; Henning, R.H.; et al. RhoA Activation Sensitizes Cells to Proteotoxic Stimuli by Abrogating the HSF1-Dependent Heat Shock Response. PLoS ONE 2015, 10, e0133553. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.; Meijering, R.A.; Hoogstra-Berends, F.; Mackovicova, K.; Vos, M.J.; Van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 2011, 6, e20395. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Zhou, S.; Guo, W.; Wang, Z.; Sun, Y.; Liu, H. SS-31 peptide enables mitochondrial targeting drug delivery: A promising therapeutic alteration to prevent hair cell damage from aminoglycosides. Drug Deliv. 2017, 24, 1750–1761. [Google Scholar] [CrossRef]
- Brundel, B.J.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef]
- Ke, L.; Qi, X.Y.; Dijkhuis, A.J.; Chartier, D.; Nattel, S.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 2008, 45, 685–693. [Google Scholar] [CrossRef]
- Brundel, B.J.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell. Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca(2+) uptake that regulates cellular metabolism. Nat. Cell. Biol. 2015, 17, 953. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SR | PAF | PeAF | LSPeAF | |
|---|---|---|---|---|
| N | 35 | 14 | 24 | 14 |
| RAA | 33 | 13 | 22 | 12 |
| LAA | 7 | 4 | 19 | 8 |
| Gender | ||||
| Male (N, %) | 26 (74.3) | 10 (71.4) | 15 (62.5) | 12 (85.7) |
| Female (N, %) | 9 (25.7) | 4 (28.6) | 9 (37.5) | 2 (14.3) |
| Age (mean ± SD) | 71 ± 12 | 70 ± 15 | 69 ± 9 | 74 ± 6 |
| Underlying heart disease (N, %) | ||||
| CAD | 24 (68.6) | 4 (28.6) | 6 (25) | 6 (42.9) |
| AVD | 2 (5.7) | 2 (14.3) | 3 (12.5) | 3 (21.4) |
| MVD | 2 (5.7) | 3 (21.4) | 11 (45.8) | 3 (21.4) |
| CAD + AVD | 6 (17.1) | 3 (21.4) | 3 (12.5) | 2 (14.3) |
| CAD + MVD | 1 (2.9) | 2 (14.3) | 1 (4.2) | 0 (0.0) |
| Duration of AF (mean ± SD (months)) | - | 89 ± 95 | 61 ± 53 | 154 ± 90 |
| LA dilatation (>45 mm, %) | 7 (20) | 3 (21.4) | 13 (54.2) | 11 (78.6) |
| LVF (N, %) | ||||
| Normal | 29 (82.9) | 11 (78.6) | 12 (50) | 9 (64.3) |
| Mild impairment | 6 (17.1) | 2 (14.3) | 6 (25) | 4 (28.6) |
| Moderate impairment | 0 (0.0) | 1 (7.1) | 5 (20.8) | 1 (7.1) |
| Severe impairment | 0 (0.0) | 0 (0.0) | 1 (4.2) | 0 (0.0) |
| Medication (N, %) | ||||
| ACE | 24 (68.6) | 8 (57.1) | 16 (66.7) | 12 (85.7) |
| Statin | 29 (82.9) | 8 (57.1) | 7 (29.2) | 10 (71.4) |
| Type I AAD | 0 (0.0) | 2 (14.3) | 0 (0.0) | 0 (0.0) |
| Type II AAD | 24 (68.6) | 7 (50) | 17 (70.8) | 12 (85.7) |
| Type III AAD | 0 (0.0) | 5 (35.7) | 3 (12.5) | 1 (7.1) |
| Type IV AAD | 0 (0.0) | 0 (0.0) | 1 (4.2) | 1 (7.1) |
| Digoxin | 0 (0.0) | 1 (7.1) | 8 (33.3) | 4 (28.6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; de Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. https://doi.org/10.3390/cells8101202
Wiersma M, van Marion DMS, Wüst RCI, Houtkooper RH, Zhang D, de Groot NMS, Henning RH, Brundel BJJM. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells. 2019; 8(10):1202. https://doi.org/10.3390/cells8101202
Chicago/Turabian StyleWiersma, Marit, Denise M.S. van Marion, Rob C.I. Wüst, Riekelt H. Houtkooper, Deli Zhang, Natasja M.S. de Groot, Robert H. Henning, and Bianca J.J.M. Brundel. 2019. "Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation" Cells 8, no. 10: 1202. https://doi.org/10.3390/cells8101202
APA StyleWiersma, M., van Marion, D. M. S., Wüst, R. C. I., Houtkooper, R. H., Zhang, D., de Groot, N. M. S., Henning, R. H., & Brundel, B. J. J. M. (2019). Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells, 8(10), 1202. https://doi.org/10.3390/cells8101202