The Incomplete Puzzle of the BCL2 Proteins


{kind=link}
{kind=link}
Abstract
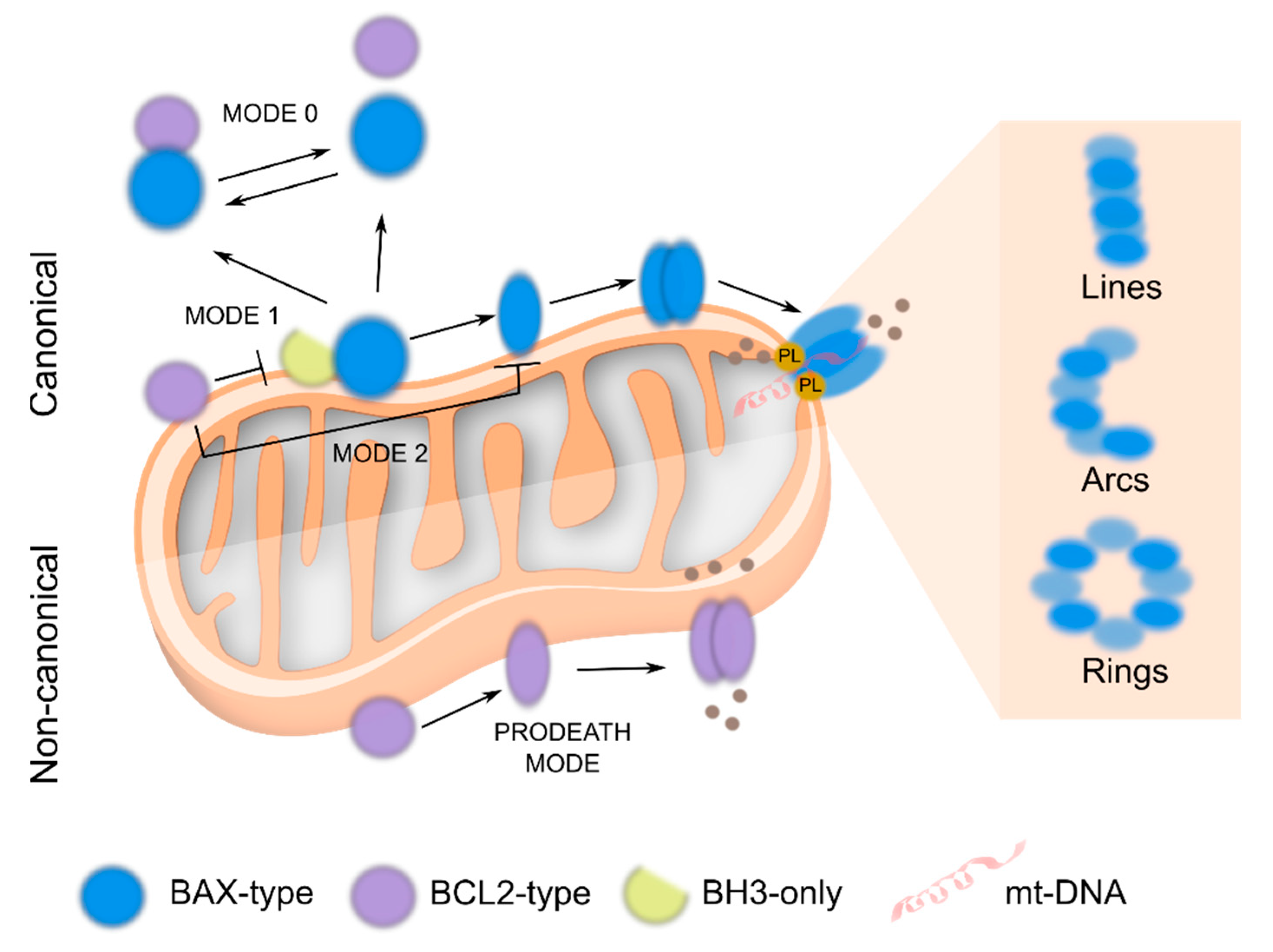
Perspective in BCL2 Universe
Acknowledgments
Funding
Conflicts of Interest
References
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2017. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.K.; Avgeris, M.; Vassilacopoulou, D.; Ardavanis, A.; Scorilas, A. Molecular Effects of Treatment of Human Colorectal Cancer Cells with Natural and Classical Chemotherapeutic Drugs: Alterations in the Expression of Apoptosis-related BCL2 Family Members, Including BCL2L12. Curr. Pharm. Biotechnol. 2018, 19, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.K.; Christodoulou, M.I.; Scorilas, A. Apoptosis-related BCL2-family members: Key players in chemotherapy. Anticancer Agents Med. Chem. 2014, 14, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Grace, C.R.; Llambi, F.; Nourse, A.; Fitzgerald, P.; Gehring, K.; Kriwacki, R.W.; Green, D.R. BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 2013, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. USA 2008, 105, 18081–18087. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Chen, H.C.; Kanai, M.; Inoue-Yamauchi, A.; Tu, H.C.; Huang, Y.; Ren, D.; Kim, H.; Takeda, S.; Reyna, D.E.; Chan, P.M.; et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 2015, 17, 1270–1281. [Google Scholar] [CrossRef]
- Leber, B.; Lin, J.; Andrews, D.W. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis Int. J. Program. Cell Death 2007, 12, 897–911. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; DuBois, G.; Haldar, S. Posttranslational modifications of Bcl2 family members—A potential therapeutic target for human malignancy. Front. Biosci. J. Virtual Libr. 2006, 11, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Kutuk, O.; Brito, G.C.; Andrews, T.S.; Leber, B.; Letai, A.; Andrews, D.W. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018, 19, e45235. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Landeta, O.; Ugarte-Uribe, B.; Cosentino, K.; Garcia-Porras, M.; Garcia-Saez, A.J.; Basanez, G. BFL1 modulates apoptosis at the membrane level through a bifunctional and multimodal mechanism showing key differences with BCLXL. Cell Death Differ. 2018, 1880–1894. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.X.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef]
- Edlich, F. The great migration of Bax and Bak. Mol. Cell. Oncol. 2015, 2, e995029. [Google Scholar] [CrossRef]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef]
- Landeta, O.; Garcia Valero, J.; Flores-Romero, H.; Bustillo-Zabalbeitia, I.; Landajuela, A.; Garcia-Porras, M.; Terrones, O.; Basanez, G. Lipid-dependent bimodal MCL1 membrane activity. ACS Chem. Biol. 2014, 9, 2852–2863. [Google Scholar] [CrossRef]
- Basanez, G.; Zhang, J.; Chau, B.N.; Maksaev, G.I.; Frolov, V.A.; Brandt, T.A.; Burch, J.; Hardwick, J.M.; Zimmerberg, J. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J. Biol. Chem. 2001, 276, 31083–31091. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kolluri, S.K.; Lin, F.; Liu, W.; Han, Y.H.; Cao, X.; Dawson, M.I.; Reed, J.C.; Zhang, X.K. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 2004, 116, 527–540. [Google Scholar] [CrossRef]
- Garcia-Saez, A.J.; Ries, J.; Orzaez, M.; Perez-Paya, E.; Schwille, P. Membrane promotes tBID interaction with BCL(XL). Nat. Struct. Mol. Biol. 2009, 16, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Bleicken, S.; Hantusch, A.; Das, K.K.; Frickey, T.; Garcia-Saez, A.J. Quantitative interactome of a membrane Bcl-2 network identifies a hierarchy of complexes for apoptosis regulation. Nat. Commun. 2017, 8, 73. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med. 2008, 8, 102–118. [Google Scholar]
- Cui, J.; Placzek, W.J. Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. Int. J. Mol. Sci. 2018, 19, 308. [Google Scholar] [CrossRef]
- Barclay, L.A.; Wales, T.E.; Garner, T.P.; Wachter, F.; Lee, S.; Guerra, R.M.; Stewart, M.L.; Braun, C.R.; Bird, G.H.; Gavathiotis, E.; et al. Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol. Cell 2015, 57, 873–886. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.C.; Kim, H.; Cheng, E.H.; Tjandra, N.; et al. BAX activation is initiated at a novel interaction site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef]
- Vasquez-Montes, V.; Vargas-Uribe, M.; Pandey, N.K.; Rodnin, M.V.; Langen, R.; Ladokhin, A.S. Lipid-modulation of membrane insertion and refolding of the apoptotic inhibitor Bcl-xL. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 691–700. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-terminal helix of Bcl-x(L) mediates Bax retrotranslocation from the mitochondria. Cell Death Diff. 2013, 20, 333–342. [Google Scholar] [CrossRef]
- Andreu-Fernandez, V.; Sancho, M.; Genoves, A.; Lucendo, E.; Todt, F.; Lauterwasser, J.; Funk, K.; Jahreis, G.; Perez-Paya, E.; Mingarro, I.; et al. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc. Natl. Acad. Sci. USA 2017, 114, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Wurm, C.A.; Bruser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; Garcia-Saez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Garcia-Saez, A.J. MAVS-induced mitochondrial membrane remodeling. FEBS J. 2019, 286, 1540–1542. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Srivastava, R.; Cao, Z.; Nedeva, C.; Naim, S.; Bachmann, D.; Rabachini, T.; Gangoda, L.; Shahi, S.; Glab, J.; Menassa, J.; et al. BCL-2 family protein BOK is a positive regulator of uridine metabolism in mammals. Proc. Natl. Acad. Sci. USA 2019, 116, 15469–15474. [Google Scholar] [CrossRef]
- Schulman, J.J.; Szczesniak, L.M.; Bunker, E.N.; Nelson, H.A.; Roe, M.W.; Wagner, L.E., 2nd; Yule, D.I.; Wojcikiewicz, R.J.H. Bok regulates mitochondrial fusion and morphology. Cell Death Diff. 2019, 826–836. [Google Scholar] [CrossRef]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.J.; Li, H.; Salvesen, G.S.; Yuan, J.; Wagner, G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 1999, 96, 615–624. [Google Scholar] [CrossRef]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Ortiz, M.; Ryan, J.; Mashaka, T.N.; Opferman, J.T.; Letai, A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2019, 391–402. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Zigart, N.; Casar, Z. A literature review of the patent publications on venetoclax - a selective Bcl-2 inhibitor: Discovering the therapeutic potential of a novel chemotherapeutic agent. Expert Opin. Ther. Pat. 2019, 29, 487–496. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Korycka-Wolowiec, A.; Wolowiec, D.; Kubiak-Mlonka, A.; Robak, T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. Expert. 2019, 15, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.A.; Clemmons, A. Venetoclax: A Novel Treatment for Patients With del(17p) Chronic Lymphocytic Leukemia. J. Adv. Pract. Oncol. 2017, 8, 647–652. [Google Scholar] [PubMed]
- Gentile, M.; Petrungaro, A.; Uccello, G.; Vigna, E.; Recchia, A.G.; Caruso, N.; Bossio, S.; De Stefano, L.; Palummo, A.; Storino, F.; et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Yecies, D.; Carlson, N.E.; Deng, J.; Letai, A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010, 115, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Paschen, S.A.; Heger, K.; Wilfling, F.; Frankenberg, T.; Bauerschmitt, H.; Seiffert, B.M.; Kirschnek, S.; Wagner, H.; Hacker, G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J. Cell Biol. 2007, 177, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Vaux, D.L. Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 2018, 25, 13–20. [Google Scholar] [CrossRef]
- Iyer, S.; Bell, F.; Westphal, D.; Anwari, K.; Gulbis, J.; Smith, B.J.; Dewson, G.; Kluck, R.M. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015, 22, 1665–1675. [Google Scholar] [CrossRef]
- Bleicken, S.; Landeta, O.; Landajuela, A.; Basanez, G.; Garcia-Saez, A.J. Proapoptotic Bax and Bak proteins form stable protein-permeable pores of tunable size. J. Biol. Chem. 2013, 288, 33241–33252. [Google Scholar] [CrossRef]
- Basanez, G.; Sharpe, J.C.; Galanis, J.; Brandt, T.B.; Hardwick, J.M.; Zimmerberg, J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J. Biol. Chem. 2002, 277, 49360–49365. [Google Scholar] [CrossRef]
- Basanez, G.; Nechushtan, A.; Drozhinin, O.; Chanturiya, A.; Choe, E.; Tutt, S.; Wood, K.A.; Hsu, Y.; Zimmerberg, J.; Youle, R.J. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl. Acad. Sci. USA. 1999, 96, 5492–5497. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, A.J.; Mingarro, I.; Perez-Paya, E.; Salgado, J. Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry 2004, 43, 10930–10943. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, A.; Smith, C.L.; Hsu, Y.T.; Youle, R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999, 18, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Yethon, J.A.; Epand, R.F.; Leber, B.; Epand, R.M.; Andrews, D.W. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 2003, 278, 48935–48941. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Westphal, D.; Dewson, G.; Ma, S.; Hockings, C.; Fairlie, W.D.; Lee, E.F.; Yao, S.; Robin, A.Y.; Smith, B.J.; et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 2013, 152, 519–531. [Google Scholar] [CrossRef]
- Cartron, P.F.; Arokium, H.; Oliver, L.; Meflah, K.; Manon, S.; Vallette, F.M. Distinct domains control the addressing and the insertion of Bax into mitochondria. J. Biol. Chem. 2005, 280, 10587–10598. [Google Scholar] [CrossRef]
- Dewson, G.; Kratina, T.; Sim, H.W.; Puthalakath, H.; Adams, J.M.; Colman, P.M.; Kluck, R.M. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 2008, 30, 369–380. [Google Scholar] [CrossRef]
- Bleicken, S.; Jeschke, G.; Stegmueller, C.; Salvador-Gallego, R.; Garcia-Saez, A.J.; Bordignon, E. Structural model of active Bax at the membrane. Mol. Cell 2014, 56, 496–505. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Garcia-Porras, M.; Basanez, G. Membrane insertion of the BAX core, but not latch domain, drives apoptotic pore formation. Sci. Rep. 2017, 7, 16259. [Google Scholar] [CrossRef] [PubMed]
- Subburaj, Y.; Cosentino, K.; Axmann, M.; Pedrueza-Villalmanzo, E.; Hermann, E.; Bleicken, S.; Spatz, J.; Garcia-Saez, A.J. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 2015, 6, 8042. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Dewson, G.; Menard, M.; Frederick, P.; Iyer, S.; Bartolo, R.; Gibson, L.; Czabotar, P.E.; Smith, B.J.; Adams, J.M.; et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, E4076–E4085. [Google Scholar] [CrossRef] [PubMed]
- Terrones, O.; Antonsson, B.; Yamaguchi, H.; Wang, H.G.; Liu, J.; Lee, R.M.; Herrmann, A.; Basanez, G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 2004, 279, 30081–30091. [Google Scholar] [CrossRef]
- Landeta, O.; Landajuela, A.; Gil, D.; Taneva, S.; Di Primo, C.; Sot, B.; Valle, M.; Frolov, V.A.; Basanez, G. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J. Biol. Chem. 2011, 286, 8213–8230. [Google Scholar] [CrossRef]
- Cheng, E.H.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Bleicken, S.; Hofhaus, G.; Ugarte-Uribe, B.; Schroder, R.; Garcia-Saez, A.J. cBid, Bax and Bcl-xL exhibit opposite membrane remodeling activities. Cell Death Dis. 2016, 7, e2121. [Google Scholar] [CrossRef]
- Valero, J.G.; Cornut-Thibaut, A.; Juge, R.; Debaud, A.L.; Gimenez, D.; Gillet, G.; Bonnefoy-Berard, N.; Salgado, J.; Salles, G.; Aouacheria, A.; et al. micro-Calpain conversion of antiapoptotic Bfl-1 (BCL2A1) into a prodeath factor reveals two distinct alpha-helices inducing mitochondria-mediated apoptosis. PLoS ONE 2012, 7, e38620. [Google Scholar] [CrossRef]
- Xie, Z.; Schendel, S.; Matsuyama, S.; Reed, J.C. Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry 1998, 37, 6410–6418. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 2009, 1788, 2080–2083. [Google Scholar] [CrossRef]
- Tamura, Y.; Endo, T.; Iijima, M.; Sesaki, H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J. Cell Biol. 2009, 185, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef] [PubMed]
- Unsay, J.D.; Cosentino, K.; Subburaj, Y.; Garcia-Saez, A.J. Cardiolipin effects on membrane structure and dynamics. Langmuir ACS J. Surf. Colloids 2013, 29, 15878–15887. [Google Scholar] [CrossRef]
- Grijalba, M.T.; Vercesi, A.E.; Schreier, S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 1999, 38, 13279–13287. [Google Scholar] [CrossRef]
- Ortiz, A.; Killian, J.A.; Verkleij, A.J.; Wilschut, J. Membrane fusion and the lamellar-to-inverted-hexagonal phase transition in cardiolipin vesicle systems induced by divalent cations. Biophys. J. 1999, 77, 2003–2014. [Google Scholar] [CrossRef]
- Kawai, F.; Shoda, M.; Harashima, R.; Sadaie, Y.; Hara, H.; Matsumoto, K. Cardiolipin domains in Bacillus subtilis marburg membranes. J. Bacteriol. 2004, 186, 1475–1483. [Google Scholar] [CrossRef]
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009, 583, 2447–2450. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Li, M.; Mandal, A.; Tyurin, V.A.; DeLucia, M.; Ahn, J.; Kagan, V.E.; van der Wel, P.C.A. Surface-Binding to Cardiolipin Nanodomains Triggers Cytochrome c Pro-apoptotic Peroxidase Activity via Localized Dynamics. Structure 2019, 27, 806–815.e4. [Google Scholar] [CrossRef]
- Aouacheria, A.; Baghdiguian, S.; Lamb, H.M.; Huska, J.D.; Pineda, F.J.; Hardwick, J.M. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 2017, 109, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef] [PubMed]
- Ugarte-Uribe, B.; Garcia-Saez, A.J. Apoptotic foci at mitochondria: In and around Bax pores. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Cleland, M.M.; Norris, K.L.; Karbowski, M.; Wang, C.; Suen, D.F.; Jiao, S.; George, N.M.; Luo, X.; Li, Z.; Youle, R.J. Bcl-2 family interaction with the mitochondrial morphogenesis machinery. Cell Death Differ. 2011, 18, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Measuring mitochondrial shape changes and their consequences on mitochondrial involvement during apoptosis. Methods Mol. Biol. 2007, 372, 405–420. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Ban, T.; Heymann, J.A.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Buggins, A.G.; Pepper, C.J. The role of Bcl-2 family proteins in chronic lymphocytic leukaemia. Leuk. Res. 2010, 34, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Straten, P.; Andersen, M.H. The anti-apoptotic members of the Bcl-2 family are attractive tumor-associated antigens. Oncotarget 2010, 1, 239–245. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Romero, H.; García-Sáez, A.J. The Incomplete Puzzle of the BCL2 Proteins. Cells 2019, 8, 1176. https://doi.org/10.3390/cells8101176
Flores-Romero H, García-Sáez AJ. The Incomplete Puzzle of the BCL2 Proteins. Cells. 2019; 8(10):1176. https://doi.org/10.3390/cells8101176
Chicago/Turabian StyleFlores-Romero, Hector, and Ana J. García-Sáez. 2019. "The Incomplete Puzzle of the BCL2 Proteins" Cells 8, no. 10: 1176. https://doi.org/10.3390/cells8101176
APA StyleFlores-Romero, H., & García-Sáez, A. J. (2019). The Incomplete Puzzle of the BCL2 Proteins. Cells, 8(10), 1176. https://doi.org/10.3390/cells8101176