TGR5 Activation Modulates an Inhibitory Effect on Liver Fibrosis Development Mediated by Anagliptin in Diabetic Rats

 , ,
, ,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. Animal Treatment
2.3. Cell Culture
2.4. Cell Proliferation Assay
2.5. RNA Extraction and Quantitative Real-Time-PCR
2.6. Protein Extraction and Western Blotting
2.7. Intrahepatic Thiobarbituric Acid Reactive Substances (TBARS) Measurement
2.8. Measurement of Cyclic AMP (cAMP) and GLP-1 Levels
2.9. Statistical Analyses
3. Results
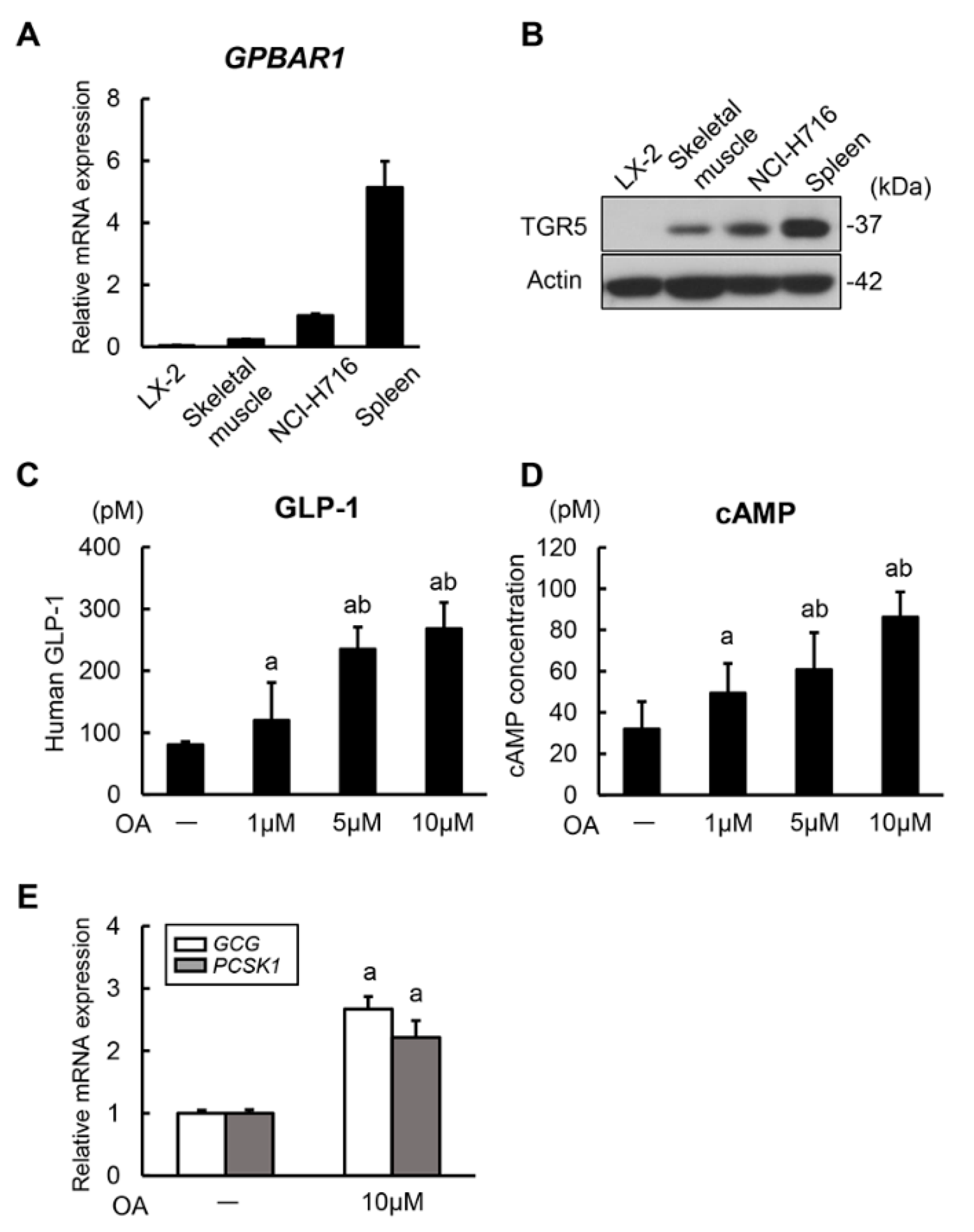
3.1. Oleanolic Acid Stimulates GLP-1 Synthesis and Secretion via TGR5 Activation in Human Intestinal Cells
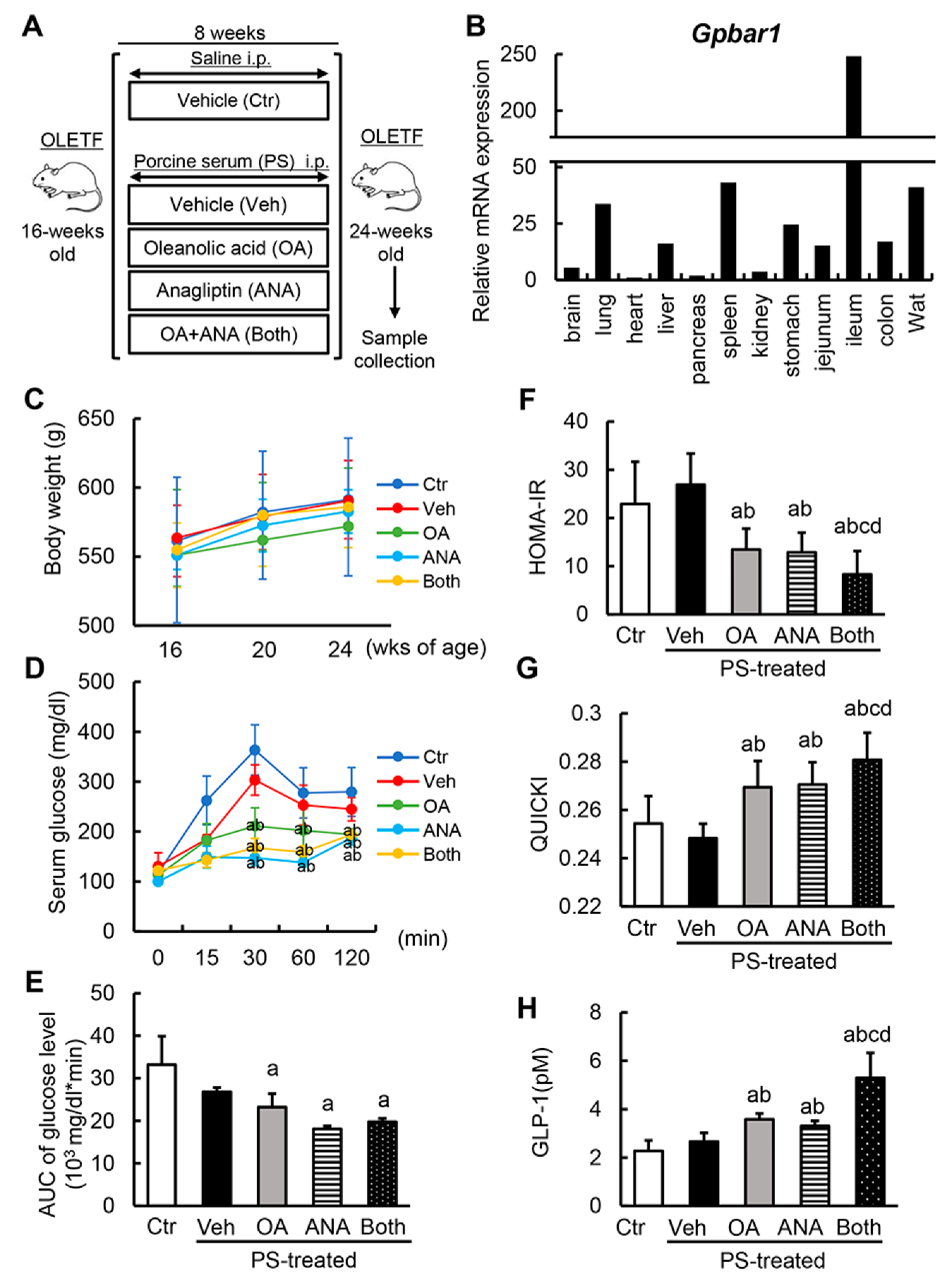
3.2. Oleanolic Acid and Anagliptin Effectively Collaborate to Exert an Antidiabetic Effect in Diabetic OLETF Rats
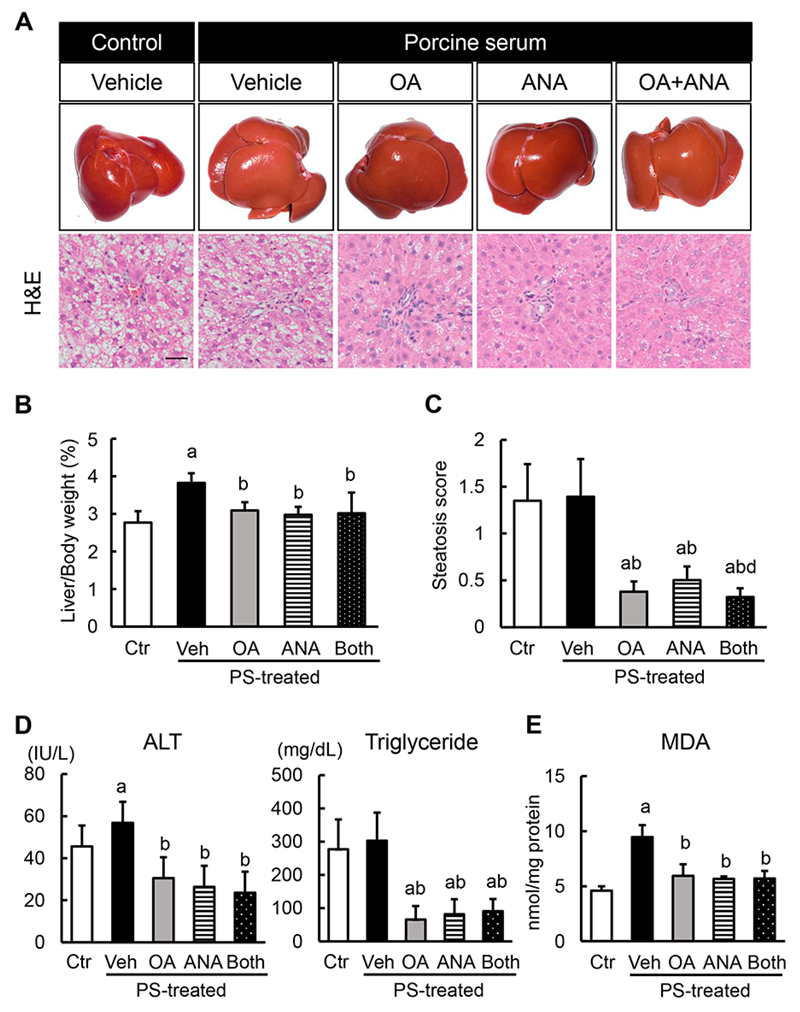
3.3. Effects of Oleanolic Acid and Anagliptin on Hepatic Steatosis and Lipid Peroxidation in OLETF Rats
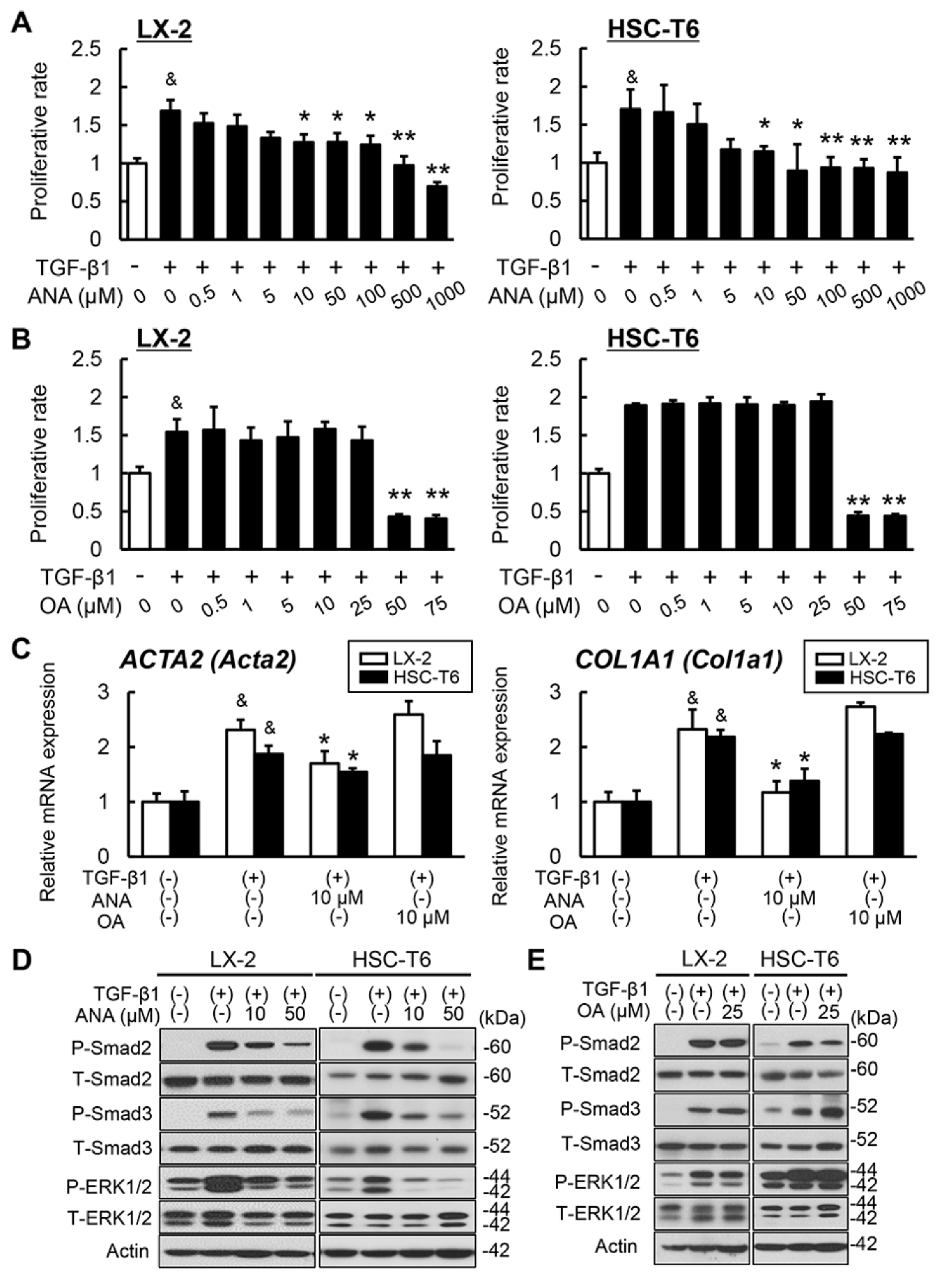
3.4. Effects of Oleanolic Acid and Anagliptin on Liver Fibrosis Development and Hepatic Stellate Cell Activation
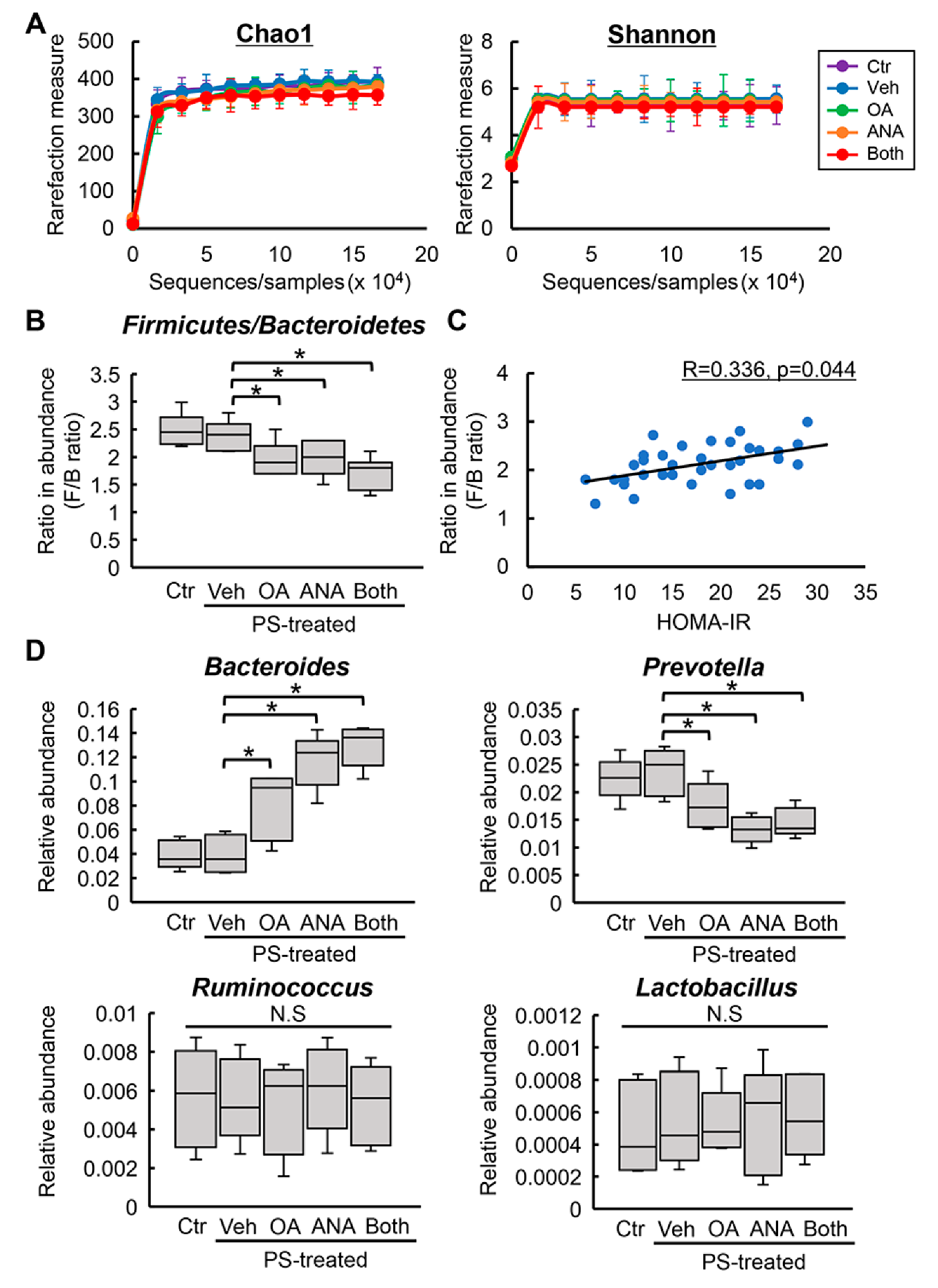
3.5. Changes in Fecal Microbial Profiles by Treatment with Oleanolic Acid and Anagliptin in OLETF Rats
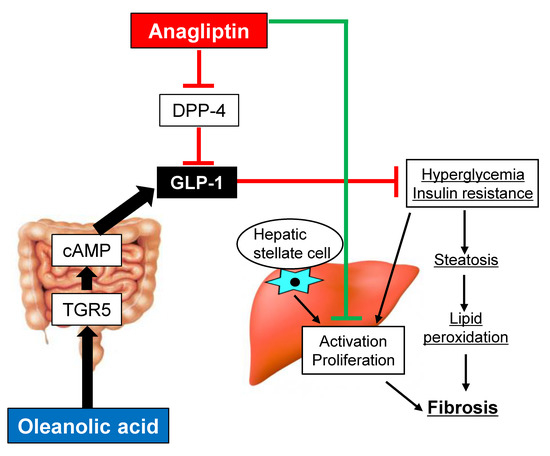
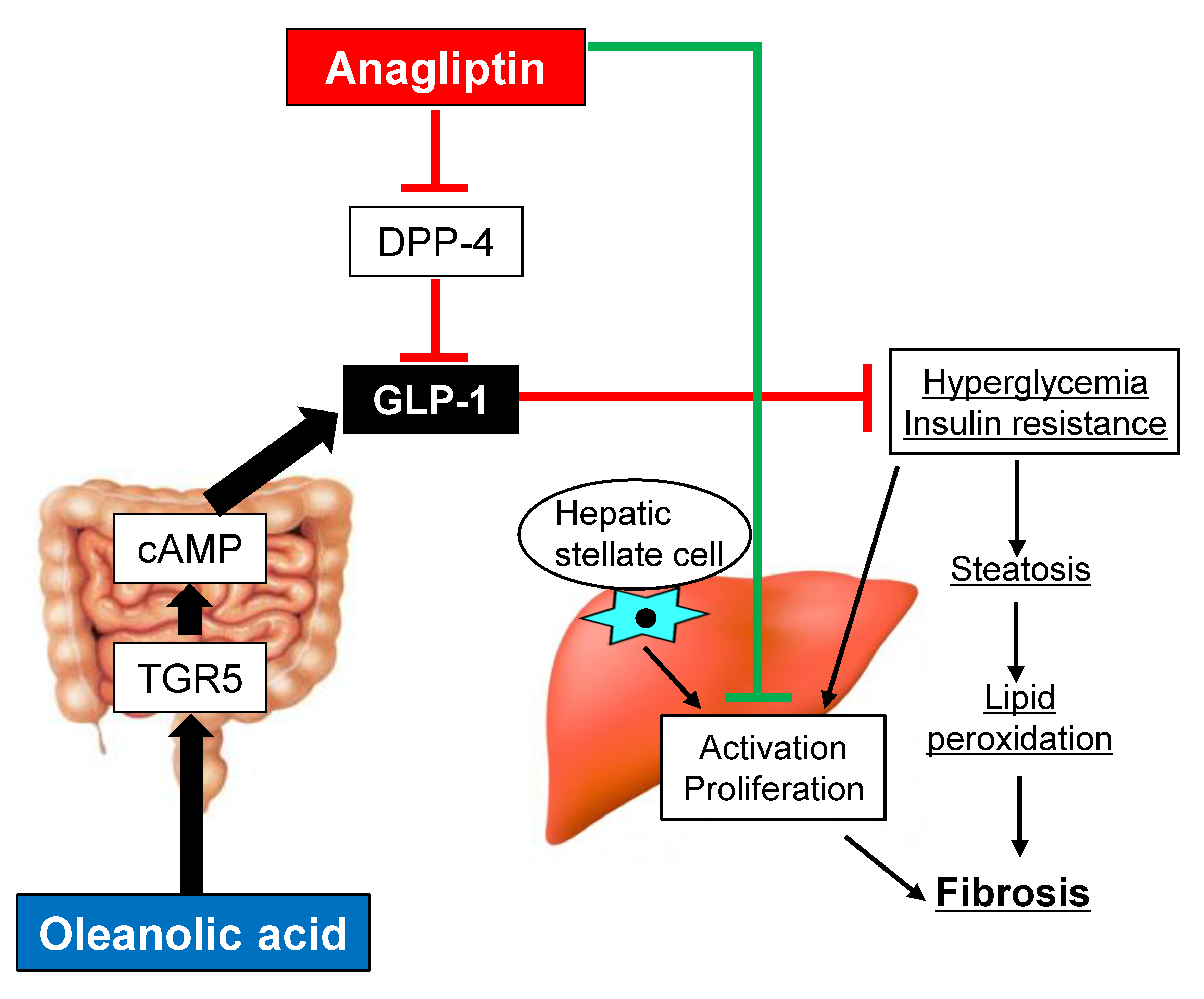
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| ANA | Anagliptin |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DPP4 | Dipeptidyl peptidase-4 |
| ERK | Extracellular signal-regulated kinase |
| FBS | Fetal bovine serum |
| GLP | Glucagon-like peptide |
| HOMA-IR | Homeostasis model assessment of insulin resistance |
| HSC | Hepatic stellate cell |
| IR | Insulin resistance |
| LETO | Long–Evans Tokushima Otsuka |
| MDA | Malondialdehyde |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| OA | Oleanolic acid |
| OGTT | Oral glucose tolerance test |
| OLETF | Otsuka Long–Evans Tokushima Fatty |
| PS | Porcine serum |
| QUICKI | Quantitative insulin sensitivity check index |
| TGR | Takeda G protein-coupled receptor |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Hepatic fibrogenesis. Semin Liver Dis. 2007, 27, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Casini, A.; Marucci, L.; Gaggiotti, G.; Orlandoni, P.; Macarri, G.; Perego, L.; Benedetti, A.; et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 1999, 29, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, R.; Enjoji, M.; Kohjima, M.; Tsuruta, S.; Fukushima, M.; Iwao, M.; Sonta, T.; Kotoh, K.; Inoguchi, T.; Nakamuta, M. High glucose stimulates hepatic stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int. 2005, 25, 1018–1026. [Google Scholar] [CrossRef]
- Petrides, A.S.; Vogt, C.; Schulze-Berge, D.; Matthews, D.; Strohmeyer, G. Pathogenesis of glucose intolerance and diabetes mellitus in cirrhosis. Hepatology 1994, 19, 616–627. [Google Scholar] [CrossRef]
- Petit, J.M.; Bour, J.B.; Galland-Jos, C.; Minello, A.; Verges, B.; Guiguet, M.; Brun, J.M.; Hillon, P. Risk factors for diabetes mellitus and early insulin resistance in chronic hepatitis C. J. Hepatol. 2001, 35, 279–283. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Yoshii, J.; Yanase, K.; Namisaki, T.; Yamazaki, M.; Moriya, K.; et al. Impact of insulin resistance on the progression of chronic liver diseases. Int. J. Mol. Med. 2008, 22, 801–808. [Google Scholar]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef]
- Kaji, K.; Yoshiji, H.; Ikenaka, Y.; Noguchi, R.; Aihara, Y.; Douhara, A.; Moriya, K.; Kawaratani, H.; Shirai, Y.; Yoshii, J.; et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J. Gastroenterol. 2014, 49, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.R.; McKenzie, C.A.; Tirona, R.G.; Summers, K.; Seney, S.; Chakrabarti, S.; Malhotra, N.; Beaton, M.D. Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J. Gastroenterol. 2017, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Tache, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Eblimit, Z.; Thevananther, S.; Karpen, S.J.; Taegtmeyer, H.; Moore, D.D.; Adorini, L.; Penny, D.J.; Desai, M.S. TGR5 activation induces cytoprotective changes in the heart and improves myocardial adaptability to physiologic, inotropic, and pressure-induced stress in mice. Cardiovasc. Ther. 2018, 36, e12462. [Google Scholar] [CrossRef] [PubMed]
- Van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Ullmer, C.; Alvarez Sanchez, R.; Sprecher, U.; Raab, S.; Mattei, P.; Dehmlow, H.; Sewing, S.; Iglesias, A.; Beauchamp, J.; Conde-Knape, K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br. J. Pharmacol. 2013, 169, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Vilsboll, T.; Christensen, M.; Junker, A.E.; Knop, F.K.; Gluud, L.L. Effects of glucagon-like peptide-1 receptor agonists on weight loss: Systematic review and meta-analyses of randomised controlled trials. BMJ 2012, 344, d7771. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Mells, J.E.; Fu, P.P.; Sharma, S.; Olson, D.; Cheng, L.; Handy, J.A.; Saxena, N.K.; Sorescu, D.; Anania, F.A. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G225–G235. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- De Bruine, A.P.; Dinjens, W.N.; van der Linden, E.P.; Pijls, M.M.; Moerkerk, P.T.; Bosman, F.T. Extracellular matrix components induce endocrine differentiation in vitro in NCI-H716 cells. Am. J. Pathol. 1993, 142, 773–782. [Google Scholar]
- Kanda, Y. Investigation of the freely available easy-to-use software ’EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423. [Google Scholar] [CrossRef]
- Rouille, Y.; Martin, S.; Steiner, D.F. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J. Biol. Chem. 1995, 270, 26488–26496. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, X.; Li, M.; Yu, M.; Ping, F.; Zheng, J.; Wang, T.; Wang, X. Vildagliptin increases butyrate-producing bacteria in the gut of diabetic rats. PLoS ONE 2017, 12, e0184735. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Sharma, A.; Arora, S.; Blomenkamp, K.; Jun, I.C.; Luong, R.; Westrich, D.J.; Mittal, A.; Buchanan, P.M.; Guzman, M.A. Preserved Gut Microbial Diversity Accompanies Upregulation of TGR5 and Hepatobiliary Transporters in Bile Acid-Treated Animals Receiving Parenteral Nutrition. JPEN J. Parenter. Enteral Nutr. 2017, 41, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Jung, Y.A.; Choi, Y.K.; Jung, G.S.; Seo, H.Y.; Kim, H.S.; Jang, B.K.; Kim, J.G.; Lee, I.K.; Kim, M.K.; Park, K.G. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res. Clin. Pract. 2014, 105, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Onoyama, T.; Koda, M.; Okamoto, T.; Kishina, M.; Matono, T.; Sugihara, T.; Murawaki, Y. Therapeutic effects of the dipeptidyl peptidase-IV inhibitor, sitagliptin, on non-alcoholic steatohepatitis in FLS-ob/ob male mice. Mol. Med. Rep. 2015, 12, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Okura, Y.; Namisaki, T.; Moriya, K.; Kitade, M.; Takeda, K.; Kaji, K.; Kim, J.G.; Lee, I.K.; Kim, M.K.; Park, K.G. Combined treatment with dipeptidyl peptidase-4 inhibitor (sitagliptin) and angiotensin-II type 1 receptor blocker (losartan) suppresses progression in a non-diabetic rat model of steatohepatitis. Hepatol. Res. 2017, 47, 1317–1328. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001, 21, 1–12. [Google Scholar] [CrossRef]
- Levy, M.T.; McCaughan, G.W.; Abbott, C.A.; Park, J.E.; Cunningham, A.M.; Muller, E.; Rettig, W.J.; Gorrell, M.D. Fibroblast activation protein: A cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology 1999, 29, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; Vieira de Ribeiro, A.J.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, D.; Wang, G.; Cui, S.; Field, R.A.; Li, J.; Zang, Y. Alogliptin alleviates liver fibrosis via suppression of activated hepatic stellate cell. Biochem. Biophys. Res. Commun. 2019, 511, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Chu, F.; Wang, M.; Li, B.; Mao, P.; Zhang, H.; Wang, P.; Guo, W.; Xu, L.; Ren, L.; et al. Ligustrazine-Oleanolic Acid Glycine Derivative, G-TOA, Selectively Inhibited the Proliferation and Induced Apoptosis of Activated HSC-T6 Cells. Molecules 2016, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Ni, H.; Chen, X.; Feng, X.; Wu, Q.; Chen, J. Identification of therapeutic effect of glucagon-like peptide 1 in the treatment of STZ-induced diabetes mellitus in rats by restoring the balance of intestinal flora. J. Cell Biochem. 2018, 119, 10067–10074. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Bose, S.; Wang, J.H.; Ansari, A.; Lim, S.K.; Chin, Y.W.; Choi, H.S.; Kim, H. Flos Lonicera Combined with Metformin Ameliorates Hepatosteatosis and Glucose Intolerance in Association with Gut Microbiota Modulation. Front. Microbiol. 2017, 8, 2271. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2-dependent and Nrf2-independent processes. Biochem Pharmacol. 2009, 77, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y.F.; Zhang, Y.; Wu, K.C.; Fan, F.; Klaassen, C.D. Oleanolic acid alters bile acid metabolism and produces cholestatic liver injury in mice. Toxicol. Appl. Pharmacol. 2013, 272, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.F.; Wan, X.L.; Xu, Y.; Liu, J. Repeated oral administration of oleanolic acid produces cholestatic liver injury in mice. Molecules 2013, 18, 3060–3071. [Google Scholar] [CrossRef] [PubMed]
- Bhunchet, E.; Eishi, Y.; Wake, K. Contribution of immune response to the hepatic fibrosis induced by porcine serum. Hepatology 1996, 23, 811–817. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaya, D.; Kaji, K.; Tsuji, Y.; Yamashita, S.; Kitagawa, K.; Ozutsumi, T.; Fujinaga, Y.; Takaya, H.; Kawaratani, H.; Moriya, K.; et al. TGR5 Activation Modulates an Inhibitory Effect on Liver Fibrosis Development Mediated by Anagliptin in Diabetic Rats. Cells 2019, 8, 1153. https://doi.org/10.3390/cells8101153
Kaya D, Kaji K, Tsuji Y, Yamashita S, Kitagawa K, Ozutsumi T, Fujinaga Y, Takaya H, Kawaratani H, Moriya K, et al. TGR5 Activation Modulates an Inhibitory Effect on Liver Fibrosis Development Mediated by Anagliptin in Diabetic Rats. Cells. 2019; 8(10):1153. https://doi.org/10.3390/cells8101153
Chicago/Turabian StyleKaya, Daisuke, Kosuke Kaji, Yuki Tsuji, Satoko Yamashita, Koh Kitagawa, Takahiro Ozutsumi, Yukihisa Fujinaga, Hiroaki Takaya, Hideto Kawaratani, Kei Moriya, and et al. 2019. "TGR5 Activation Modulates an Inhibitory Effect on Liver Fibrosis Development Mediated by Anagliptin in Diabetic Rats" Cells 8, no. 10: 1153. https://doi.org/10.3390/cells8101153
APA StyleKaya, D., Kaji, K., Tsuji, Y., Yamashita, S., Kitagawa, K., Ozutsumi, T., Fujinaga, Y., Takaya, H., Kawaratani, H., Moriya, K., Namisaki, T., Akahane, T., & Yoshiji, H. (2019). TGR5 Activation Modulates an Inhibitory Effect on Liver Fibrosis Development Mediated by Anagliptin in Diabetic Rats. Cells, 8(10), 1153. https://doi.org/10.3390/cells8101153