Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer

 ,
,  ,
, 


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
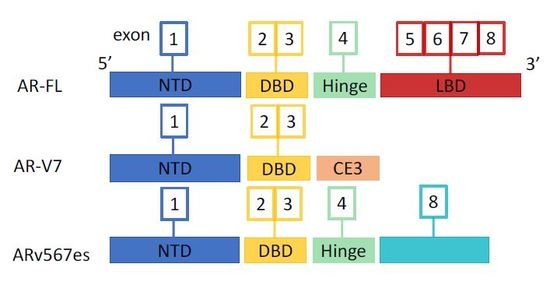
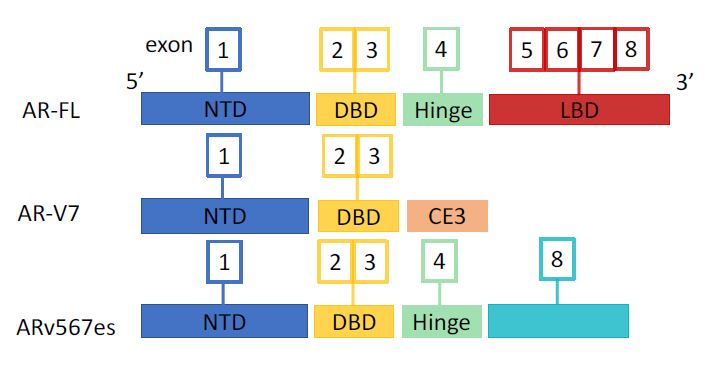
2. Androgen Receptor and Splice Variants
3. Mechanisms of Resistance to Therapies
4. Epigenetic Mechanisms
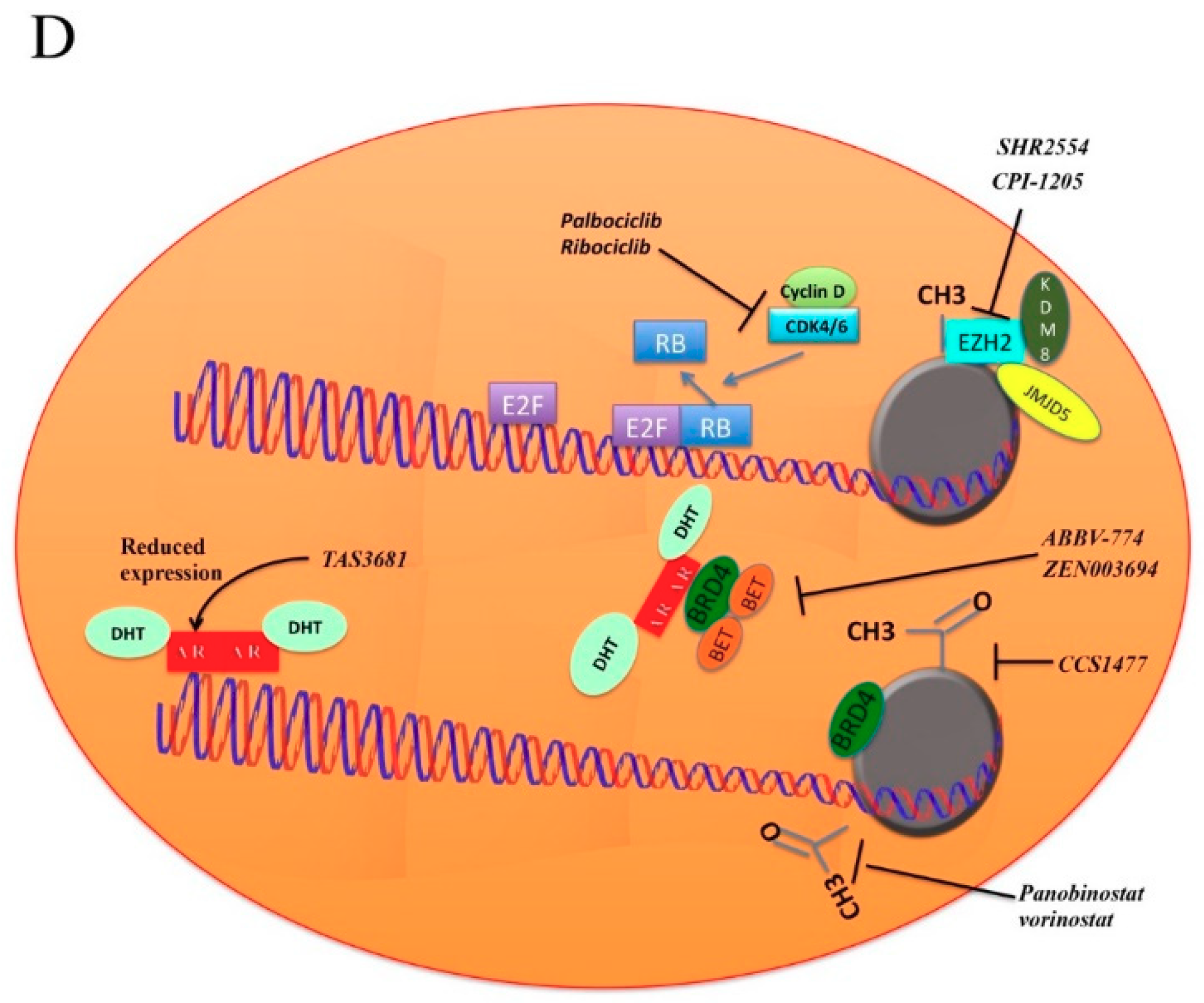
5. How to Overcome Resistance to AR Inhibition
5.1. BET Pathway
5.2. Cyclins
5.3. Histone Deacetylase Inhibitors
5.4. EZH2
5.5. PI3K-AKT-mTOR
5.6. SRC
5.7. Other Strategies to Overcome Hormone Inhibition Resistance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PCa | Prostate Cancer |
| ADT | Androgen Deprivation Therapy |
| CRPC | Castration Resistant Prostate Cancer |
| AR | Androgen Receptor |
| AR-FL | Full-Length AR |
| NTD | N-Terminal Domain |
| DBD | DNA-Binding Domain |
| LBD | Ligand Binding Domain |
| AR-Vs | AR splice Variants |
| DNMTs | DNA-methyltransferases |
| HATs | Histone Acetyltransferases |
| HDACs | Histone Deacetylases |
| CTCs | Circulating Tumor Cells |
| CYP17A1 | Cytochrome P450 family 17 subfamily A polypeptide 1 |
| CYP11A1 | Cytochrome P450 family 11 subfamily A polypeptide 1 |
| DHEA | Dehydroepiandrosterone |
| DHEA-S | Dehydroepiandrosterone sulfate |
| DHT | Dihydrotestosterone |
| Akt | Protein kinase B |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| mTOR | Mammalian target of rapamycin |
| EZH2 | Enhancer of zeste homolog 2 |
| BRD4 | Bromodomain containing protein 4 |
| BET | Bromodomain and extra terminal family proteins |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Wilt, T.J.; Jones, K.M.; Barry, M.J.; Andriole, G.L.; Culkin, D.; Wheeler, T.; Aronson, W.J.; Brawer, M.K. Follow-up of prostatectomy versus observation for early prostate cancer. N. Engl. J. Med. 2017, 377, 132–142. [Google Scholar] [CrossRef]
- Bill-Axelson, A.; Holmberg, L.; Garmo, H.; Rider, J.R.; Taari, K.; Busch, C.; Nordling, S.; Häggman, M.; Andersson, S.O.; Bratell, S.; et al. Radical prostatectomy or watchful waiting in early prostate cancer. N. Engl. J. Med. 2014, 370, 932–942. [Google Scholar] [CrossRef]
- Parker, C. Active surveillance: Towards a new paradigm in the management of early prostate cancer. Lancet Oncol. 2004, 5, 101–106. [Google Scholar] [CrossRef]
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef]
- Donovan, J.L.; Hamdy, F.C.; Lane, J.A.; Mason, M.; Metcalfe, C.; Walsh, E.; Blazeby, J.M.; Peters, T.J.; Holding, P.; Bonnington, S.; et al. Patient-reported outcomes after monitoring, surgery, or radiotherapy for prostate cancer. N. Engl. J. Med. 2016, 375, 1425–1437. [Google Scholar] [CrossRef]
- Mason, M.D.; Parulekar, W.R.; Sydes, M.R.; Brundage, M.; Kirkbride, P.; Gospodarowicz, M.; Cowan, R.; Kostashuk, E.C.; Anderson, J.; Swanson, G.; et al. Final report of the intergroup randomized study of combined androgen-deprivation therapy plus radiotherapy versus androgen-deprivation therapy alone in locally advanced prostate cancer. J. Clin. Oncol. 2015, 33, 2143–2150. [Google Scholar] [CrossRef]
- Messing, E.M.; Manola, J.; Yao, J.; Kiernan, M.; Crawford, D.; Wilding, G.; di’SantAgnese, P.A.; Trump, D. Eastern Cooperative Oncology Group study EST 3886. Immediate versus deferred androgen deprivation treatment in patients with node-positive prostate cancer after radical prostatectomy and pelvic lymphadenectomy. Lancet Oncol. 2006, 7, 472–479. [Google Scholar] [CrossRef]
- Klotz, L. Maximal androgen blockade for advanced prostate cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 331–340. [Google Scholar] [CrossRef]
- Samson, D.J.; Seidenfeld, J.; Schmitt, B.; Hasselblad, V.; Albertsen, P.C.; Bennett, C.L.; Wilt, T.J.; Aronson, N. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer 2002, 95, 361–376. [Google Scholar] [CrossRef]
- Gravis, G.; Boher, J.M.; Joly, F.; Soulie, M.; Albiges, L.; Priou, F.; Latorzeff, I.; Delva, R.; Krakowski, I.; Laguerre, B.; et al. Androgen Deprivation Therapy (ADT) plus docetaxel versus ADT alone in metastatic non castrate prostate cancer: Impact of metastatic burden and long-term survival analysis of the randomized phase 3 GETUG-AFU15 Trial. Eur. Urol. 2016, 70, 256–262. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal therapy in metastatic hormone- sensitive prostate cancer: Long-term survival analysis of the randomized phase III E3805 CHAARTED trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Rydzewska, L.H.M.; Burdett, S.; Vale, C.L.; Clarke, N.W.; Fizazi, K.; Kheoh, T.; Mason, M.D.; Miladinovic, B.; James, N.D.; Parmar, M.K.B.; et al. Adding abiraterone to androgen deprivation therapy in men with metastatic hormone-sensitive prostate cancer: A systematic review and meta-analysis. Eur. J. Cancer 2017, 84, 88–101. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef]
- Massari, F.; Di Nunno, V.; Comito, F.; Cubelli, M.; Ciccarese, C.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Ardizzoni, A. Circulating tumor cells in genitourinary tumors. Ther. Adv. Urol. 2017, 10, 65–77. [Google Scholar] [CrossRef]
- Di Nunno, V.; Gatto, L.; Santoni, M.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Scarpelli, M.; Montironi, R.; Massari, F. Recent Advances in Liquid Biopsy in Patients with Castration Resistant Prostate Cancer. Front. Oncol. 2018, 8, 397. [Google Scholar] [CrossRef]
- Ciccarese, C.; Santoni, M.; Brunelli, M.; Buti, S.; Modena, A.; Nabissi, M.; Artibani, W.; Martignoni, G.; Montironi, R.; Tortora, G.; et al. AR-V7 and prostate cancer: The watershed for treatment selection? Cancer Treat. Rev. 2016, 43, 27–35. [Google Scholar] [CrossRef]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; de Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–667. [Google Scholar] [CrossRef]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Nava Rodrigues, D.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2018, 129. [Google Scholar] [CrossRef]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Bitting, R.L.; Schaeffer, D.; Somarelli, J.A.; Garcia-Blanco, M.A.; Armstrong, A.J. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014, 33, 441–468. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Tan, J.; Sharief, Y.; Hamil, K.G.; Gregory, C.W.; Zang, D.Y.; Sar, M.; Gumerlock, P.H.; deVere White, R.W.; Pretlow, T.G.; Harris, E.; et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol. Endocrinol. 1997, 11, 450–459. [Google Scholar] [CrossRef]
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar]
- Zhao, X.Y.; Malloy, P.J.; Krishnan, A.V.; Swami, S.; Navone, N.M.; Peehl, D.M.; Feldman, D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat. Med. 2000, 6, 703–706. [Google Scholar] [CrossRef]
- Van de Wijngaart, D.J.; Moller, M.; Lusher, S.J.; Hersmus, R.; Jenster, G.; Trapman, J.; Dubbink, H.J. Systematic structure- function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J. Biol. Chem. 2010, 285, 5097–5105. [Google Scholar] [CrossRef]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full- length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef]
- Cimadamore, A.; Gasparrini, S.; Scarpelli, M.; Doria, A.; Mazzucchelli, R.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Montironi, R. Epigenetic modifications and modulators in prostate cancer. Crit. Rev. Oncog. 2017, 22, 439–450. [Google Scholar] [CrossRef]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef]
- Geybels, M.S.; Zhao, S.; Wong, C.J.; Bibikova, M.; Klotzle, B.; Wu, M.; Ostrander, E.A.; Fan, J.B.; Feng, Z.; Stanford, J.L. Epigenomic profiling of DNA methylation in paired prostate cancer versus adjacent benign tissue. Prostate 2015, 75, 1941–1950. [Google Scholar] [CrossRef]
- Zhao, S.; Geybels, M.S.; Leonardson, A.; Rubicz, R.; Kolb, S.; Yan, Q.; Klotzle, B.; Bibikova, M.; Hurtado-Coll, A.; Troyer, D.; et al. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men diagnosed with clinically localized prostate cancer. Clin. Cancer Res. 2017, 23, 311–319. [Google Scholar] [CrossRef]
- Vanaja, D.K.; Ehrich, M.; Van den Boom, D.; Cheville, J.C.; Karnes, R.J.; Tindall, D.J.; Cantor, C.R.; Young, C.Y. Hypermethylation of genes for diagnosis and risk stratification of prostate cancer. Cancer Investig. 2009, 27, 549–560. [Google Scholar] [CrossRef]
- Kwan, P.S.; Lau, C.C.; Chiu, Y.T.; Man, C.; Liu, J.; Tang, K.D.; Wong, Y.C.; Ling, M.T. Daxx regulates mitotic progression and prostate cancer predisposition. Carcinogenesis 2013, 34, 750–759. [Google Scholar] [CrossRef]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef]
- Bhaumik, S.R.; Smith, E.; Shilatifard, A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007, 14, 1008–1016. [Google Scholar] [CrossRef]
- Jia, L.; Shen, H.C.; Wantroba, M.; Khalid, O.; Liang, G.; Wang, Q.; Gentzschein, E.; Pinski, J.K.; Stanczyk, F.Z.; Jones, P.A.; et al. Locus-wide chromatin remodeling and enhanced androgen receptor-mediated transcription in recurrent prostate tumor cells. Mol. Cell. Biol. 2006, 26, 7331–7341. [Google Scholar] [CrossRef]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol. 2011, 2011, 862125. [Google Scholar] [CrossRef]
- Fu, M.; Liu, M.; Sauve, A.A.; Jiao, X.; Zhang, X.; Wu, X.; Powell, M.J.; Yang, T.; Gu, W.; Avantaggiati, M.L.; et al. Hormonal control of androgen receptor function through SIRT1. Mol. Cell. Biol. 2006, 26, 8122–8135. [Google Scholar] [CrossRef]
- Damodaran, S.; Damaschke, N.; Gawdzik, J.; Yang, B.; Shi, C.; Allen, G.O.; Huang, W.; Denu, J.; Jarrard, D. Dysregulation of Sirtuin 2 (SIRT2) and histone H3K18 acetylation pathways associates with adverse prostate cancer out- comes. BMC Cancer 2017, 17, 874. [Google Scholar] [CrossRef]
- Karanikolas, B.D.; Figueiredo, M.L.; Wu, L. Polycomb group protein enhancer of zeste 2 is an oncogene that promotes the neoplastic transformation of a benign prostatic epithelial cell line. Mol. Cancer Res. 2009, 7, 1456–1465. [Google Scholar] [CrossRef]
- Shiogama, S.; Yoshiba, S.; Soga, D.; Motohashi, H.; Shintani, S. Aberrant expression of EZH2 is associated with pathological findings and P53 alteration. Anticancer Res. 2013, 33, 4309–4317. [Google Scholar]
- Cimadamore, A.; Gasparrini, S.; Santoni, M.; Cheng, L.; Lopez-Beltran, A.; Battelli, N.; Massari, F.; Giunchi, F.; Fiorentino, M.; Scarpelli, M.; et al. Biomarkers of aggressiveness in genitourinary tumors with emphasis on kidney, bladder and prostate cancer. Exp. Rev. Mol. Diagn. 2018, 18, 645–655. [Google Scholar] [CrossRef]
- Labbè, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadosky, K.M.; Ku, S.Y.; Sjöström, M.; Alshalalfa, M.; Erho, N.; et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef]
- Wang, H.J.; Pochampalli, M.; Wang, L.Y.; Zou, J.X.; Li, P.S.; Hsu, S.C.; Wang, B.J.; Huang, S.H.; Yang, P.; Yang, J.C.; et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2018. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Merkle, D.; Hoffmann, R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: Cross-talk with the androgen receptor. Cell Signal. 2011, 23, 507–515. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Batra, A.; Winquist, E. Emerging cell cycle inhibitors for treating metastatic castration-resistant prostate cancer. Expert. Opin. Emerg. Drugs 2018, 23, 271–282. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.; Ju, X.; Vetuschi, A.; Sferra, R.; Casimiro, M.; Pompili, S.; Festuccia, C.; Colapietro, A.; Gaudio, E.; et al. Cyclin D1 silencing suppresses tumorigenicity, impairs DNA double strand break repair and thus radiosensitizes androgen-independent prostate cancer cells to DNA damage. Oncotarget 2016, 7, 5383–5400. [Google Scholar] [CrossRef]
- Pal, S.K.; Patel, J.; He, M.; Foulk, B.; Kraft, K.; Smirnov, D.A.; Twardowski, P.; Kortylewski, M.; Bhargava, V.; Jones, J.O. Identification of mechanisms of resistance to treatment with abiraterone acetate or enzalutamide in patients with castration-resistant prostate cancer (CRPC). Cancer 2018, 124, 1216–1224. [Google Scholar] [CrossRef]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Alumkal, J.J.; Stein, M.N.; Taplin, M.E.; Babb, J.; Barnett, E.S.; Gomez-Pinillos, A.; Liu, X.; Moore, D.; DiPaola, R.; et al. Epigenetic Therapy with Panobinostat Combined with Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Nava Rodrigues, D.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Jiao, J.; Wang, S.; Qiao, R.; Vivanco, I.; Watson, P.A.; Sawyers, C.L.; Wu, H. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007, 67, 6083–6091. [Google Scholar] [CrossRef]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef]
- Szafran, A.T.; Stephan, C.; Bolt, M.; Mancini, M.G.; Marcelli, M.; Mancini, M.A. High-Content Screening Identifies Src Family Kinases as Potential Regulators of AR-V7 Expression and Androgen-Independent Cell Growth. Prostate 2017, 77, 82–93. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Wang, J.; Qin, M.; Gao, L.; Holtz, R.; Vessella, R.L.; Leach, R.W.; Gelman, I.H. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2017, 8, 10324–10347. [Google Scholar] [CrossRef]
- Kajiwara, D.; Minamiguchi, K.; Seki, M.; Mizutani, H.; Yamamura, K.; Okajima, S.; Sasaki, E.; Utsugi, T.; Iwasawa, Y. The impact of TAS3681, a new type of androgen receptor antagonist, on aberrant AR signaling that drives tumor resistance to AR-targeted therapies by down-regulating full length and splice variant AR. J. Clin. Oncol. 2017, 35, 197. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells 2019, 8, 43. https://doi.org/10.3390/cells8010043
Mollica V, Di Nunno V, Cimadamore A, Lopez-Beltran A, Cheng L, Santoni M, Scarpelli M, Montironi R, Massari F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells. 2019; 8(1):43. https://doi.org/10.3390/cells8010043
Chicago/Turabian StyleMollica, Veronica, Vincenzo Di Nunno, Alessia Cimadamore, Antonio Lopez-Beltran, Liang Cheng, Matteo Santoni, Marina Scarpelli, Rodolfo Montironi, and Francesco Massari. 2019. "Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer" Cells 8, no. 1: 43. https://doi.org/10.3390/cells8010043
APA StyleMollica, V., Di Nunno, V., Cimadamore, A., Lopez-Beltran, A., Cheng, L., Santoni, M., Scarpelli, M., Montironi, R., & Massari, F. (2019). Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells, 8(1), 43. https://doi.org/10.3390/cells8010043