The Role of Lipids in Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
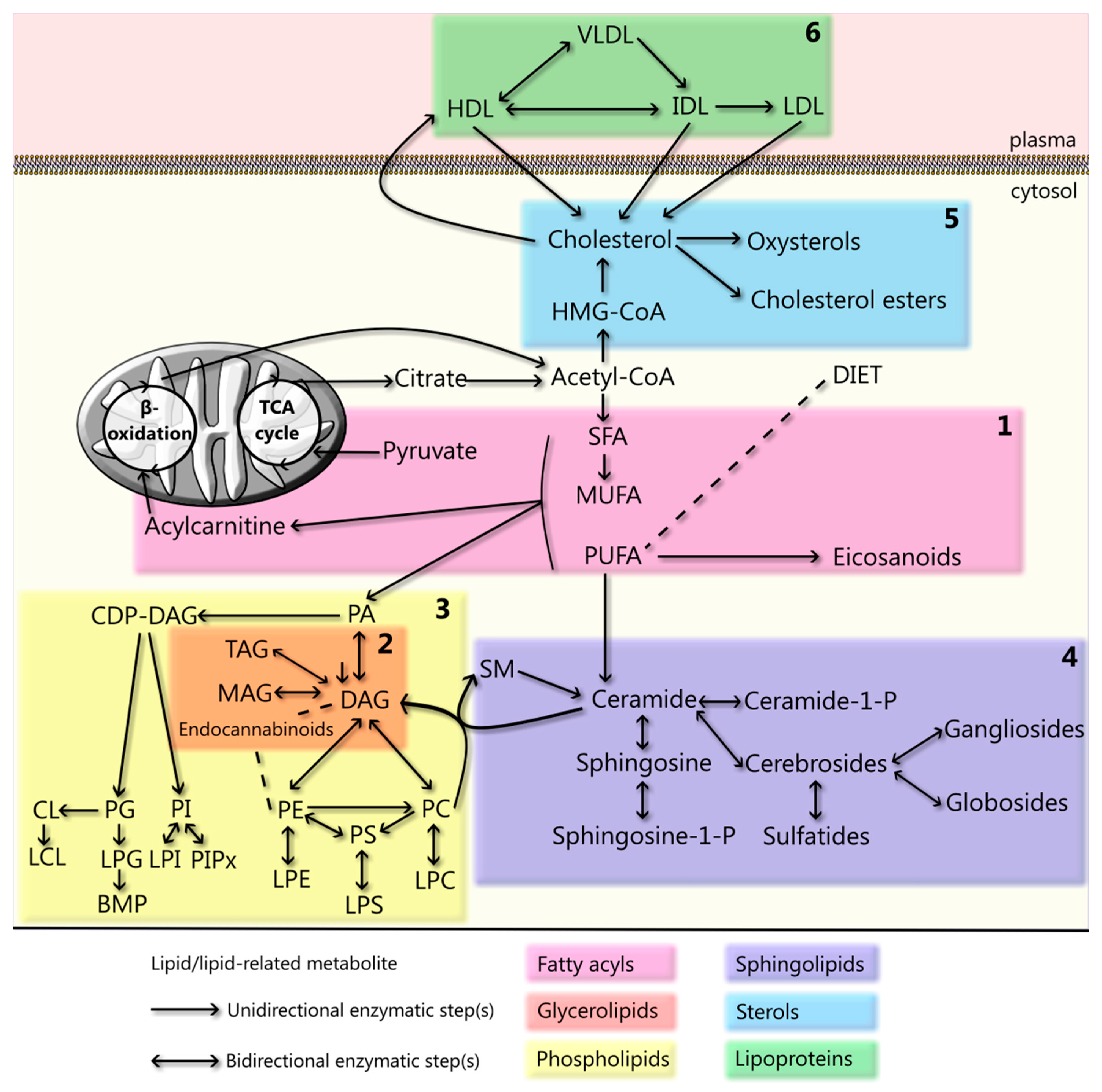
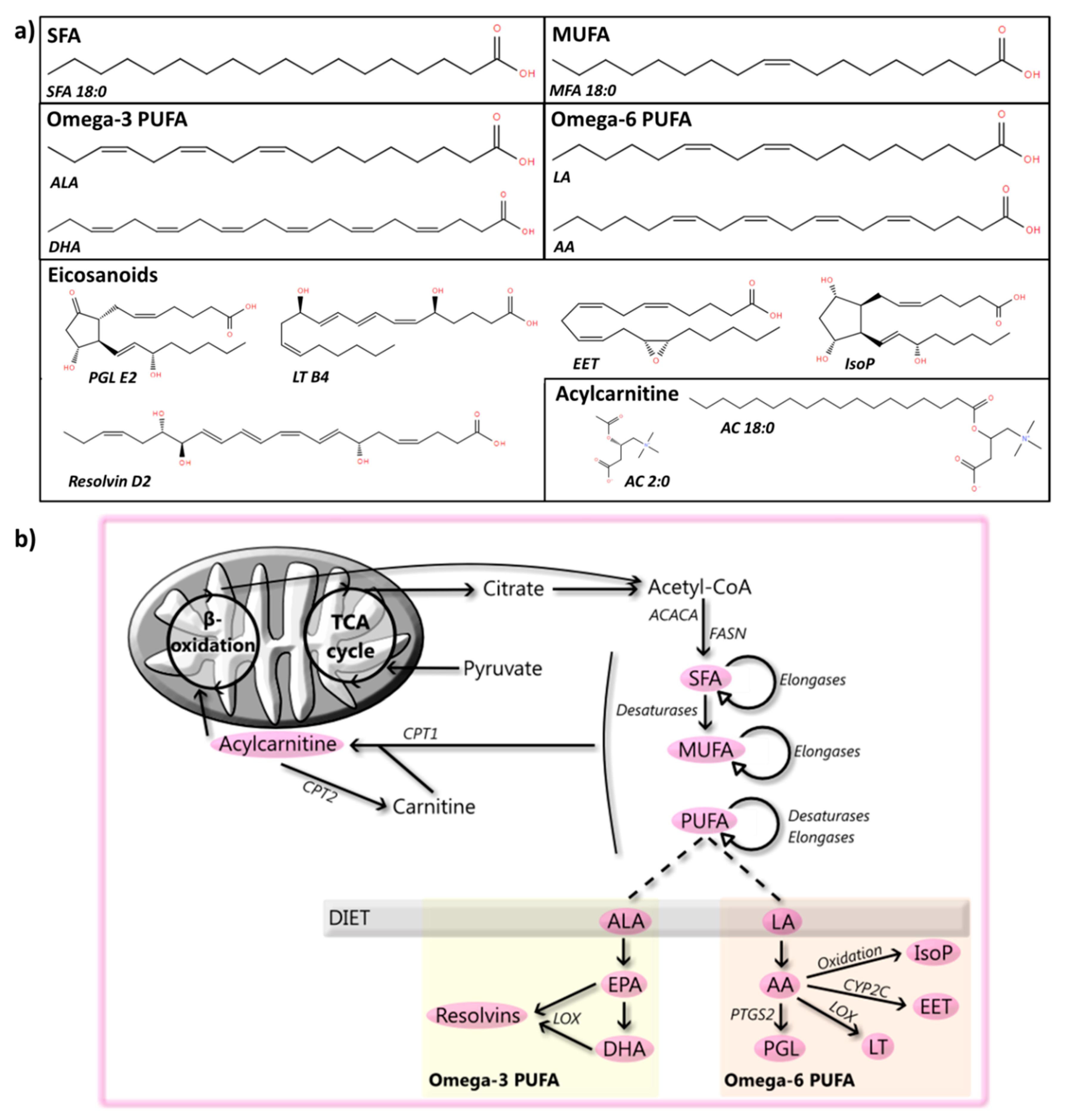
2. Fatty Acyls
2.1. SFA
2.2. MUFA
2.3. PUFA
2.3.1. Human Studies on PUFA
2.3.2. Animal and Cellular Studies on PUFA
2.3.3. Alpha-Synuclein and PUFA
2.4. Eicosanoids and Docosanoids
2.4.1. PGL
2.4.2. LT
2.4.3. EET
2.4.4. Isoprostanes
2.4.5. Other Eicosanoids and Docosanoids
2.5. Carnitine and Acylcarnitine
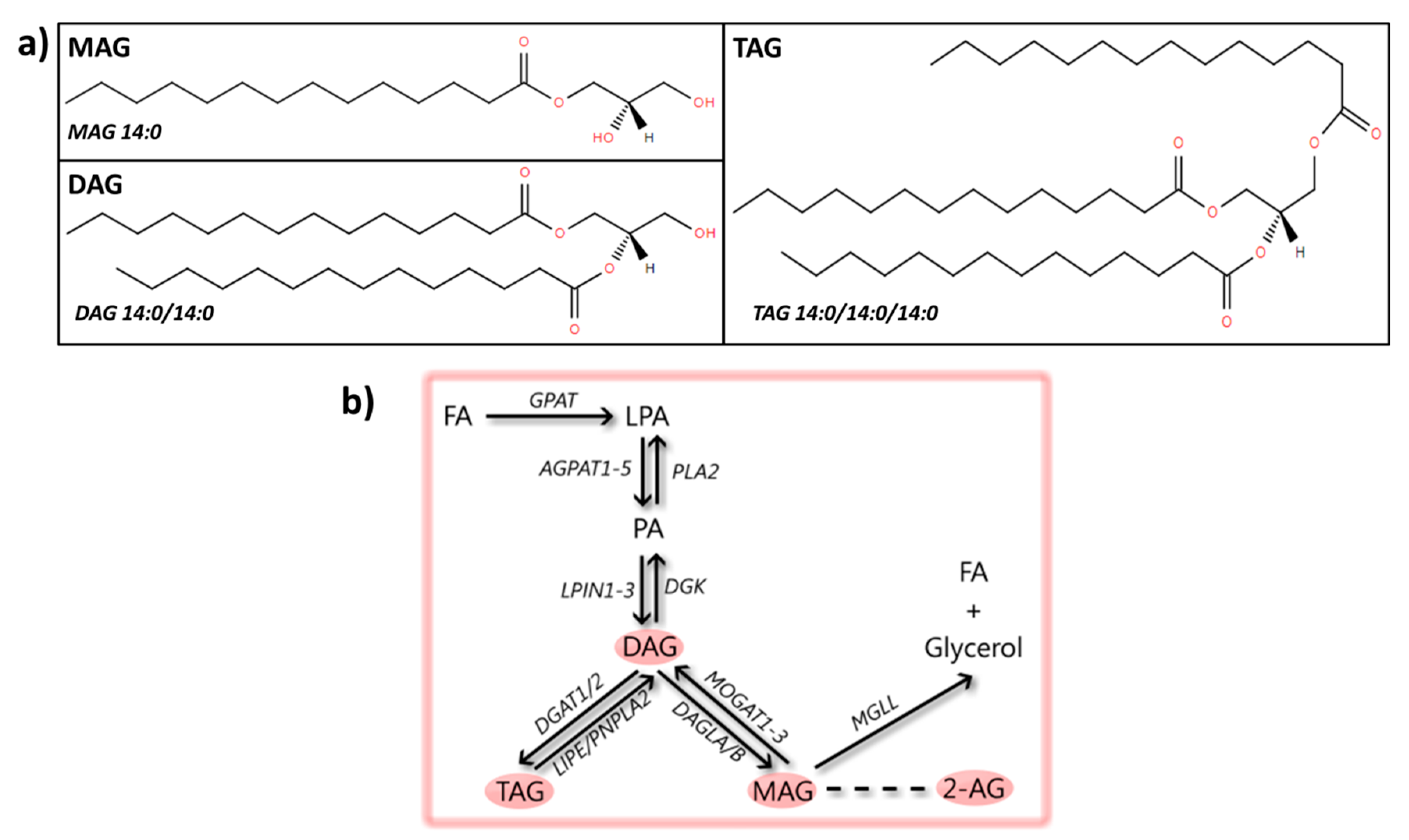
3. Glycerolipids
3.1. MAG
Endocannabinoids
3.2. DAG
3.3. TAG
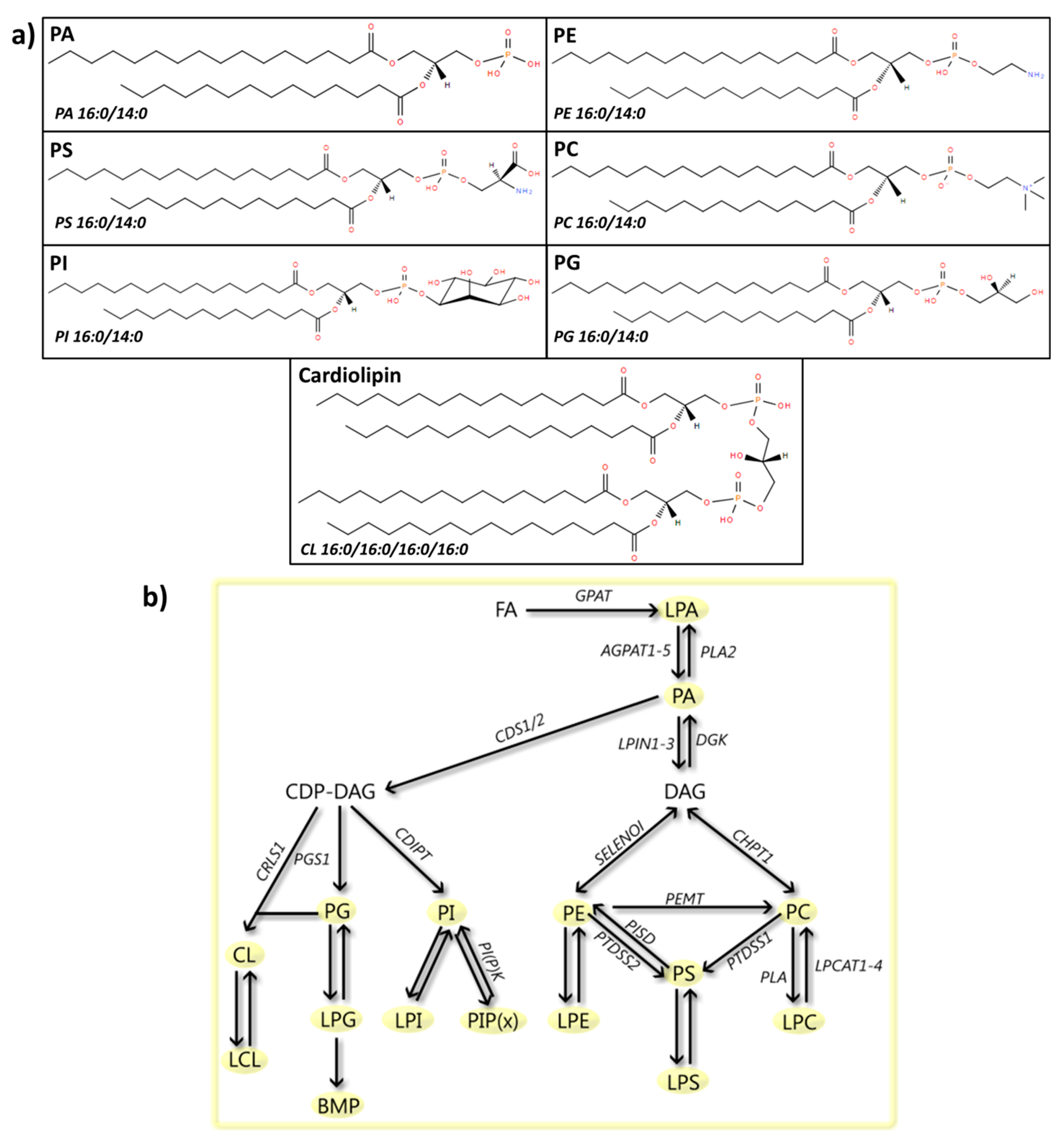
4. Glycerophospholipids
4.1. PA
LPA
4.2. PE
4.3. PS
4.4. PC
LPC
4.5. PI
PI Phosphate (PIPx)
4.6. PG
4.7. CL
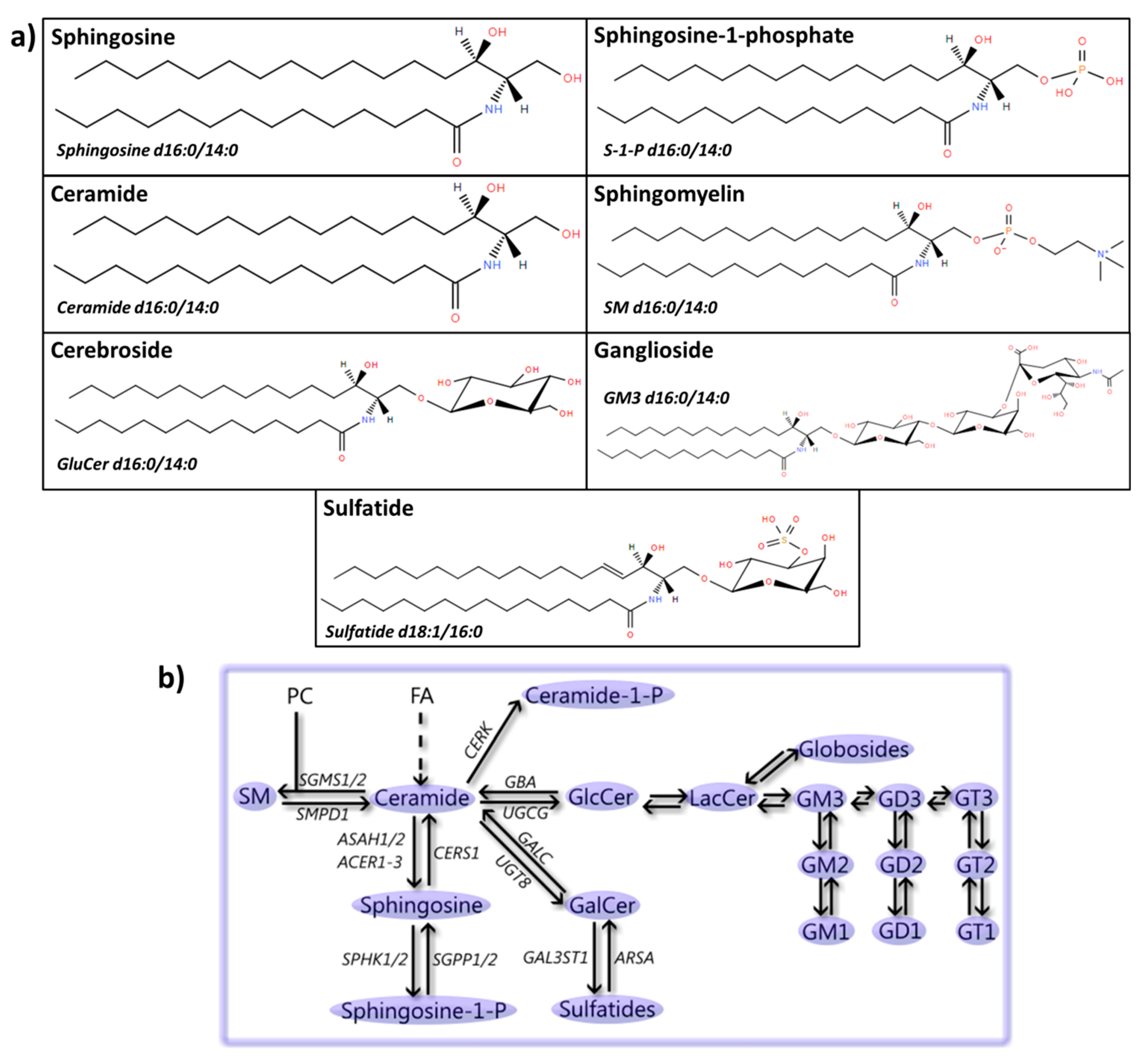
5. Sphingolipids
5.1. Sphingosine(-1-Phosphate)
5.2. Ceramide
5.3. SM
5.4. Cerebrosides
5.5. Gangliosides
5.6. Sulfatides
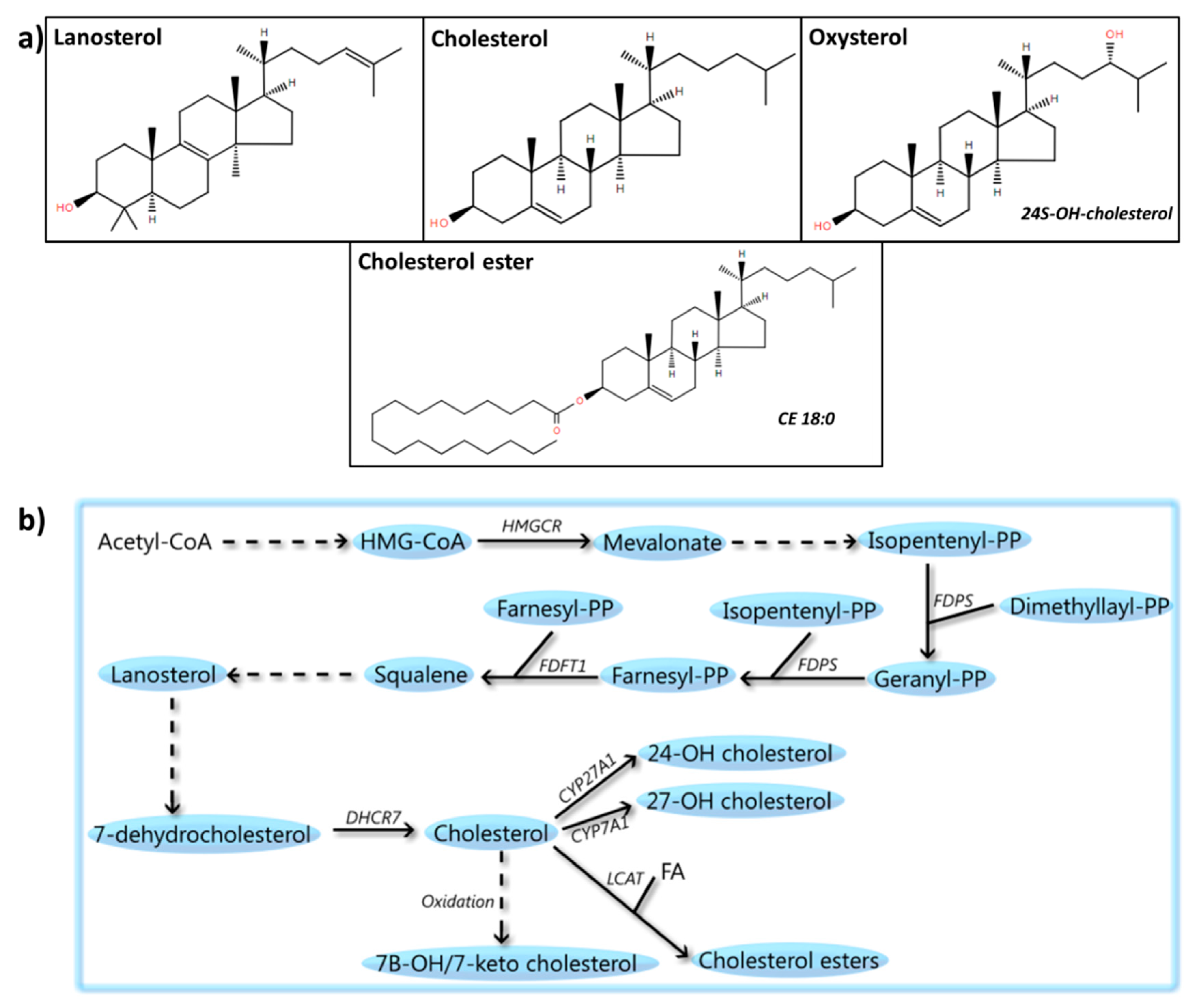
6. Sterols
6.1. Cholesterol
6.1.1. Human Studies on Cholesterol
6.1.2. Animal and Cellular Studies on Cholesterol
6.1.3. Alpha-Synuclein and Cholesterol
6.1.4. Statins
6.2. Cholesterol Precursors
6.3. CEs
6.4. Oxysterols
7. Lipoproteins
7.1. HDL
7.2. LDL
7.3. VLDL
8. The Cellular Lipidome
9. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Mao, Z.H. Progression of motor symptoms in Parkinson’s disease. Neurosci. Bull. 2012, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Przedborski, S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30, 475–483. [Google Scholar] [CrossRef]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef]
- Nichols, W.C.; Pankratz, N.; Marek, D.K.; Pauciulo, M.W.; Elsaesser, V.E.; Halter, C.A.; Rudolph, A.; Wojcieszek, J.; Pfeiffer, R.F.; Foround, T.; et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009, 72, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Landston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, N.; Wilk, J.B.; Latourelle, J.C.; DeStefano, A.L.; Halter, C.; Pugh, E.W.; Doheny, K.F.; Gausella, J.F.; Nichols, W.C.; Foroud, T.; et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 2009, 124, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Nalls, M.A.; Plagnol, V.; et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef]
- Horton, T.J.; Drougas, H.; Brachey, A.; Reed, G.W.; Peters, J.C.; Hill, J.O. Fat and carbohydrate overfeeding in humans: Different effects on energy storage. Am. J. Clin. Nutr. 1995, 62, 19–29. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Fernandis, A.Z.; Wenk, M.R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 2007, 18, 121–128. [Google Scholar] [CrossRef]
- Bieberich, E. It’s a lipid’s world: Bioactive lipid metabolism and signaling in neural stem cell differentiation. Neurochem. Res. 2012, 37, 1208–1229. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, D.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Essential Fatty acids—A review. Curr. Pharm. Biotechnol. 2006, 7, 467–482. [Google Scholar] [CrossRef]
- Calder, P.C. Functional Roles of Fatty Acids and Their Effects on Human Health. JPEN J. Parenter Enteral Nutr. 2015, 39, 18S–32S. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S. [Google Scholar] [CrossRef]
- Tan, L.C.; Methawasin, K.; Tan, E.K.; Tan, J.H.; Au, W.L.; Yuan, J.M.; Koh, W.P. Dietary cholesterol, fats and risk of Parkinson’s disease in the Singapore Chinese Health Study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 86–92. [Google Scholar] [CrossRef]
- Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary fat intake and risk of Parkinson’s disease: A case-control study in Japan. J. Neurol. Sci. 2010, 288, 117–122. [Google Scholar] [CrossRef]
- De Lau, L.M.L.; Bornebroek, M.; Witteman, J.C.M.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M.B. Dietary fatty acids and the risk of Parkinson disease: The Rotterdam study. Neurology 2005, 64, 2040–2045. [Google Scholar] [CrossRef]
- Kamel, F.; Goldman, S.M.; Umbach, D.M.; Chen, H.; Richardson, G.; Barber, M.R.; Meng, C.; Marras, C.; Koerll, M.; Kasten, M.; et al. Dietary fat intake, pesticide use, and Parkinson’s disease. Park. Relat. Disord. 2014, 20, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Berthiaume, L.; Hadj-Tahar, A.; Rajput, A.H.; Bédard, P.J.; Di Paolo, T.; Julien, P.; Calon, F. Postmortem brain fatty acid profile of levodopa-treated Parkinson disease patients and parkinsonian monkeys. Neurochem. Int. 2006, 48, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Virtue, S.; Xu, X.; Tan, C.Y.; Griffin, J.L.; Miller, A.K.; Vidal-Puig, A.; Teleman, A.A. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 2015, 525, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Bajracharya, R.; Bustamante, S.; Ballard, J.W.O. Stearic acid supplementation in high protein to carbohydrate (P:C) ratio diet improves physiological and mitochondrial functions of Drosophila melanogaster parkin null mutants. J. Gerontol. A Biol. Sci. Med. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bajracharya, R.; Ballard, J.W.O. Dietary management and physical exercise can improve climbing defects and mitochondrial activity in Drosophila melanogaster parkin null mutants. Fly 2018, 12, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Joniec-Maciejak, I.; Wawer, A.; Turzyńska, D.; Sobolewska, A.; Maciejak, P.; Szyndler, J.; Mirowska-Guzel, D.; Płaźnik, A. Octanoic acid prevents reduction of striatal dopamine in the MPTP mouse model of Parkinson’s disease. Pharmacol. Rep. 2018, 70, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.W.; Say, Y.H. Palmitic acid induces neurotoxicity and gliatoxicity in SH-SY5Y human neuroblastoma and T98G human glioblastoma cells. PeerJ 2018, 6, e4696. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Fuente-Martin, E.; Finan, B.; Kim, M.; Frank, A.; Garcia-Caceres, C.; Navas, C.; Gordillo, R.; Neinast, M.; Kalainayakan, S.P.; et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chu, Y.; Kordower, J.H.; Li, B.; Cao, H.; Huang, L.; Nishida, M.; Song, L.; Wang, D.; Federoff, H.J. PGC-1α Promoter Methylation in Parkinson’s Disease. PLoS ONE 2015, 10, e0134087. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Leenders, K.L. Cyclooxygenase and neuroinflammation in Parkinson’s disease neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Faergeman, N.J.; Cole, N.B.; Castagnet, P.I.; Nussbaum, R.L.; Murphy, E.J. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding. Biochemistry 2005, 44, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.P.; Schleicher, E.D.; Cegan, A.; Deuschle, C.; Baur, S.; Hauser, A.K.; Synofzik, M.; Srulijes, K.; Brockmann, K.; Berg, D.; et al. Cerebrospinal fluid fatty acids in glucocerebrosidase-associated Parkinson’s disease. Mov. Disord. 2012, 27, 288–292. [Google Scholar] [CrossRef]
- Heller, A.; Won, L.; Bubula, N.; Hessefort, S.; Kurutz, J.W.; Reddy, G.A.; Gross, M. Long-chain fatty acids increase cellular dopamine in an immortalized cell line (MN9D) derived from mouse mesencephalon. Neurosci. Lett. 2005, 376, 35–39. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; De Luca, R.; Mazur, K.; Chepkova, A.N.; Haas, H.L.; Bauer, A. N-oleoyldopamine modulates activity of midbrain dopaminergic neurons through multiple mechanisms. Neuropharmacology 2017, 119, 111–122. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef]
- Kubo, S.; Nemani, V.M.; Chalkley, R.J.; Anthony, M.D.; Hattori, N.; Mizuno, Y.; Edwards, R.H.; Fortin, D.L. A combinatorial code for the interaction of alpha-synuclein with membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar] [CrossRef] [PubMed]
- Tvrzicka, E.; Kremmyda, L.S.; Stankova, B.; Zak, A. Fatty acids as biocompounds: Their role in human metabolism, health and disease—A review. Part 1: Classification, dietary sources and biological functions. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2011, 155, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Raphael, W.; Sordillo, L.M. Dietary polyunsaturated fatty acids and inflammation: The role of phospholipid biosynthesis. Int. J. Mol. Sci. 2013, 14, 21167–21188. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Lillycrop, K.A. Fatty acids and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.I.F.F.; Kiliaan, A.J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Beard, J.D.; Umbach, D.M.; Park, Y.; Huang, X.; Blair, A.; Kamel, F.; Chen, H. Dietary fat intake and risk for Parkinson’s disease. Mov. Disord. 2014, 29, 1623–1630. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Willett, W.C.; Ascherio, A. Dietary intakes of fat and risk of Parkinson’s disease. Am. J. Epidemiol. 2003, 157, 1007–1014. [Google Scholar] [CrossRef]
- Schulte, E.C.; Altmaier, E.; Berger, H.S.; Do, K.T.; Kastenmüller, G.; Wahl, S.; Adamski, J.; Peters, A.; Krumsiek, J.; Suhre, K.; et al. Alterations in Lipid and Inositol Metabolisms in Two Dopaminergic Disorders. PLoS ONE 2016, 11, e0147129. [Google Scholar] [CrossRef]
- Selley, M.L. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson’s disease. Free Radic. Biol. Med. 1998, 25, 169–174. [Google Scholar] [CrossRef]
- Abbott, S.K.; Jenner, A.M.; Spiro, A.S.; Batterham, M.; Halliday, G.M.; Garner, B. Fatty acid composition of the anterior cingulate cortex indicates a high susceptibility to lipid peroxidation in Parkinson’s disease. J. Park. Dis. 2015, 5, 175–185. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Saint-Pierre, M.; Julien, C.; Salem, N.; Cicchetti, F.; Calon, F. Beneficial effects of dietary omega-3 polyunsaturated fatty acid on toxin-induced neuronal degeneration in an animal model of Parkinson’s disease. FASEB J. 2008, 22, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Gibrat, C.; Saint-Pierre, M.; Julien, C.; Calon, F.; Cicchetti, F. Modulation of brain-derived neurotrophic factor as a potential neuroprotective mechanism of action of omega-3 fatty acids in a parkinsonian animal model. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Barros, A.S.; Crispim, R.Y.G.; Cavalcanti, J.U.; Souza, R.B.; Lemos, J.C.; Cristino Filho, G.; Bezerra, M.M.; Pinheiro, T.F.M.; de Vasconcelos, S.M.M.; Macêdo, D.S.; et al. Impact of the Chronic Omega-3 Fatty Acids Supplementation in Hemiparkinsonism Model Induced by 6-Hydroxydopamine in Rats. Basic Clin. Pharmacol. Toxicol. 2017, 120, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wang, J.; Li, M.; Liu, Q.; Wei, D.; Yang, M.; Kong, L. (1)H NMR-based metabolomics study on a goldfish model of Parkinson’s disease induced by 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP). Chem. Biol. Interact. 2014, 223, 18–26. [Google Scholar] [CrossRef]
- Cardoso, H.D.; dos Santos Junior, E.F.; de Santana, D.F.; Gonçalves-Pimentel, C.; Angelim, M.K.; Isaac, A.R.; Lagranha, C.J.; Guedes, R.C.; Beltrão, E.I.; Morya, E.; et al. Omega-3 deficiency and neurodegeneration in the substantia nigra: Involvement of increased nitric oxide production and reduced BDNF expression. Biochim. Biophys. Acta 2014, 1840, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Delattre, A.M.; Carabelli, B.; Mori, M.A.; Kempe, P.G.; Rizzo de Souza, L.E.; Zanata, S.M.; Machado, R.B.; Suchecki, D.; Andrade da Costa, B.L.S.; Lima, M.M.S.; et al. Maternal Omega-3 Supplement Improves Dopaminergic System in Pre- and Postnatal Inflammation-Induced Neurotoxicity in Parkinson’s Disease Model. Mol. Neurobiol. 2017, 54, 2090–2106. [Google Scholar] [CrossRef]
- Tanriover, G.; Seval-Celik, Y.; Ozsoy, O.; Akkoyunlu, G.; Savcioglu, F.; Hacioglu, G.; Demir, N.; Agar, A. The effects of docosahexaenoic acid on glial derived neurotrophic factor and neurturin in bilateral rat model of Parkinson’s disease. Folia Histochem. Cytobiol. 2010, 48, 434–441. [Google Scholar] [CrossRef]
- Hacioglu, G.; Seval-Celik, Y.; Tanriover, G.; Ozsoy, O.; Saka-Topcuoglu, E.; Balkan, S.; Agar, A. Docosahexaenoic acid provides protective mechanism in bilaterally MPTP-lesioned rat model of Parkinson’s disease. Folia Histochem. Cytobiol. 2012, 50, 228–238. [Google Scholar] [CrossRef]
- Ozkan, A.; Parlak, H.; Tanriover, G.; Dilmac, S.; Ulker, S.N.; Birsen, I.; Agar, A. The protective mechanism of docosahexaenoic acid in mouse model of Parkinson: The role of hemeoxygenase. Neurochem. Int. 2016, 101, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Han, J.; Jang, Y.; Kim, S.J.; Park, J.H.; Seo, K.S.; Jeong, S.; Shin, S.; Lim, K.; Heo, J.Y.; et al. Docosahexaenoic acid prevents paraquat-induced reactive oxygen species production in dopaminergic neurons via enhancement of glutathione homeostasis. Biochem. Biophys. Res. Commun. 2015, 457, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Serrano-García, N.; Fernández-Valverde, F.; Luis-Garcia, E.R.; Granados-Rojas, L.; Juárez-Zepeda, T.E.; Orozco-Suárez, S.A.; Pedraza-Chaverri, J.; Orozco-Ibarra, M.; Jiménez-Anguiano, A. Docosahexaenoic acid protection in a rotenone induced Parkinson’s model: Prevention of tubulin and synaptophysin loss, but no association with mitochondrial function. Neurochem. Int. 2018, 121, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Shashikumar, S.; Pradeep, H.; Chinnu, S.; Rajini, P.S.; Rajanikant, G.K. Alpha-linolenic acid suppresses dopaminergic neurodegeneration induced by 6-OHDA in C. elegans. Physiol. Behav. 2015, 151, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, K.; Saint-Pierre, M.; Cisbani, G.; St-Amour, I.; Gibrat, C.; Giguère-Rancourt, A.; Calon, F.; Cicchetti, F. Partial neurorescue effects of DHA following a 6-OHDA lesion of the mouse dopaminergic system. J. Nutr. Biochem. 2016, 30, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Hernando, S.; Requejo, C.; Herran, E.; Ruiz-Ortega, J.A.; Morera-Herreras, T.; Lafuente, J.V.; Ugedo, L.; Gainza, E.; Pedraz, J.L.; Igartua, M.; et al. Beneficial effects of n-3 polyunsaturated fatty acids administration in a partial lesion model of Parkinson’s disease: The role of glia and NRf2 regulation. Neurobiol. Dis. 2019, 121, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Soler, M.; Cordobilla, B.; Morató, X.; Fernández-Dueñas, V.; Domingo, J.C.; Ciruela, F. Triglyceride Form of Docosahexaenoic Acid Mediates Neuroprotection in Experimental Parkinsonism. Front. Neurosci. 2018, 12, 604. [Google Scholar] [CrossRef]
- Chang, Y.L.; Chen, S.J.; Kao, C.L.; Hung, S.C.; Ding, D.C.; Yu, C.C.; Chen, Y.J.; Ku, H.H.; Lin, C.P.; Lee, K.H.; et al. Docosahexaenoic acid promotes dopaminergic differentiation in induced pluripotent stem cells and inhibits teratoma formation in rats with Parkinson-like pathology. Cell Transplant. 2012, 21, 313–332. [Google Scholar] [CrossRef]
- Parlak, H.; Ozkan, A.; Dilmac, S.; Tanriover, G.; Ozsoy, O.; Agar, A. Neuronal nitric oxide synthase phosphorylation induced by docosahexaenoic acid protects dopaminergic neurons in an experimental model of Parkinson’s disease. Folia Histochem. Cytobiol. 2015, 56, 27–37. [Google Scholar] [CrossRef]
- Luchtman, D.W.; Meng, Q.; Wang, X.; Shao, D.; Song, C. ω-3 fatty acid eicosapentaenoic acid attenuates MPP+-induced neurodegeneration in fully differentiated human SH-SY5Y and primary mesencephalic cells. J. Neurochem. 2013, 124, 855–868. [Google Scholar] [CrossRef]
- Meng, Q.; Luchtman, D.W.; El Bahh, B.; Zidichouski, J.A.; Yang, J.; Song, C. Ethyl-eicosapentaenoate modulates changes in neurochemistry and brain lipids induced by parkinsonian neurotoxin 1-methyl-4-phenylpyridinium in mouse brain slices. Eur. J. Pharmacol. 2010, 649, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Luchtman, D.W.; Meng, Q.; Song, C. Ethyl-eicosapentaenoate (E-EPA) attenuates motor impairments and inflammation in the MPTP-probenecid mouse model of Parkinson’s disease. Behav. Brain Res. 2012, 226, 386–396. [Google Scholar] [CrossRef]
- Mori, M.A.; Delattre, A.M.; Carabelli, B.; Pudell, C.; Bortolanza, M.; Staziaki, P.V.; Visentainer, J.V.; Montanher, P.F.; Del Bel, E.A.; Ferraz, A.C. Neuroprotective effect of omega-3 polyunsaturated fatty acids in the 6-OHDA model of Parkinson’s disease is mediated by a reduction of inducible nitric oxide synthase. Nutr. Neurosci. 2018, 21, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Delattre, A.M.; Kiss, A.; Szawka, R.E.; Anselmo-Franci, J.A.; Bagatini, P.B.; Xavier, L.L.; Rigon, P.; Achaval, M.; Iagher, F.; de David, C.; et al. Evaluation of chronic omega-3 fatty acids supplementation on behavioral and neurochemical alterations in 6-hydroxydopamine-lesion model of Parkinson’s disease. Neurosci. Res. 2010, 66, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Kabuto, H.; Amakawa, M.; Mankura, M.; Yamanushi, T.T.; Mori, A. Docosahexaenoic acid ethyl ester enhances 6-hydroxydopamine-induced neuronal damage by induction of lipid peroxidation in mouse striatum. Neurochem. Res. 2009, 34, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Shamoto-Nagai, M.; Hisaka, S.; Naoi, M.; Maruyama, W. Modification of α-synuclein by lipid peroxidation products derived from polyunsaturated fatty acids promotes toxic oligomerization: Its relevance to Parkinson disease. J. Clin. Biochem. Nutr. 2018, 62, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Abramov, A.Y.; Shchepinov, M.S. Lipid peroxidation is essential for α-synuclein-induced cell death. J. Neurochem. 2015, 133, 582–589. [Google Scholar] [CrossRef]
- Shchepinov, M.S.; Chou, V.P.; Pollock, E.; Langston, J.W.; Cantor, C.R.; Molinari, R.J.; Manning-Boğ, A.B. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson’s disease. Toxicol. Lett. 2011, 207, 97–103. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138 Pt 7, 1801–1816. [Google Scholar] [CrossRef]
- Denny Joseph, K.M.; Muralidhara. Combined oral supplementation of fish oil and quercetin enhances neuroprotection in a chronic rotenone rat model: Relevance to Parkinson’s disease. Neurochem. Res. 2015, 40, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Bazinet, R.P.; Rapoport, S.I.; Bhattacharjee, A.K. Brain arachidonic acid cascade enzymes are upregulated in a rat model of unilateral Parkinson disease. Neurochem. Res. 2010, 35, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Chalimoniuk, M.; Stolecka, A.; Ziemińska, E.; Stepień, A.; Langfort, J.; Strosznajder, J.B. Involvement of multiple protein kinases in cPLA2 phosphorylation, arachidonic acid release, and cell death in in vivo and in vitro models of 1-methyl-4-phenylpyridinium-induced parkinsonism—The possible key role of PKG. J. Neurochem. 2009, 110, 307–317. [Google Scholar] [CrossRef]
- Tang, K.S. Protective effect of arachidonic acid and linoleic acid on 1-methyl-4-phenylpyridinium-induced toxicity in PC12 cells. Lipids Health Dis. 2014, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Yabuki, Y.; Kobayashi, Y.; Onozato, M.; Owada, Y.; Fukunaga, K. FABP3 protein promotes α-synuclein oligomerization associated with 1-methyl-1, 2, 3, 6-tetrahydropiridine-induced neurotoxicity. J. Biol. Chem. 2014, 289, 18957–18965. [Google Scholar] [CrossRef]
- Wang, Y.; Plastina, P.; Vincken, J.P.; Jansen, R.; Balvers, M.; Ten Klooster, J.P.; Gruppen, H.; Witkamp, R.; Meijerink, J. N-Docosahexaenoyl Dopamine, an Endocannabinoid-like Conjugate of Dopamine and the n-3 Fatty Acid Docosahexaenoic Acid, Attenuates Lipopolysaccharide-Induced Activation of Microglia and Macrophages via COX-2. ACS Chem. Neurosci. 2017, 8, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. Alpha-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 2009, 10, 218–234. [Google Scholar] [CrossRef]
- Darios, F.; Ruipérez, V.; López, I.; Villanueva, J.; Gutierrez, L.M.; Davletov, B. Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010, 11, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Karube, H.; Sakamoto, M.; Arawaka, S.; Hara, S.; Sato, H.; Ren, C.H.; Goto, S.; Koyama, S.; Wada, M.; Kawanami, T.; et al. N-terminal region of alpha-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett. 2008, 582, 3693–3700. [Google Scholar] [CrossRef]
- Israeli, E.; Sharon, R. Beta-synuclein occurs in vivo in lipid-associated oligomers and forms hetero-oligomers with alpha-synuclein. J. Neurochem. 2009, 108, 465–474. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Assayag, K.; Yakunin, E.; Loeb, V.; Selkoe, D.J.; Sharon, R. Polyunsaturated fatty acids induce alpha-synuclein-related pathogenic changes in neuronal cells. Am. J. Pathol. 2007, 171, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Broersen, K.; van den Brink, D.; Fraser, G.; Goedert, M.; Davletov, B. Alpha-synuclein adopts an alpha-helical conformation in the presence of polyunsaturated fatty acids to hinder micelle formation. Biochemistry 2006, 45, 15610–15616. [Google Scholar] [CrossRef] [PubMed]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. A-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson’s disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhen, J.; Lu, Z. Synergetic Neuroprotective Effect of Docosahexaenoic Acid and Aspirin in SH-Y5Y by Inhibiting miR-21 and Activating RXRα and PPARα. DNA Cell Biol. 2017, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, G.; Frare, E.; Pivato, M.; Relini, A.; Penco, A.; Greggio, E.; Bubacco, L.; Fontana, A.; de Laureto, P.P. Structural and morphological characterization of aggregated species of α-synuclein induced by docosahexaenoic acid. J. Biol. Chem. 2011, 286, 22262–22274. [Google Scholar] [CrossRef]
- Fecchio, C.; De Franceschi, G.; Relini, A.; Greggio, E.; Dalla Serra, M.; Bubacco, L.; de Laureto, P.P. α-Synuclein oligomers induced by docosahexaenoic acid affect membrane integrity. PLoS ONE 2013, 8, e82732. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Bubacco, L.; Mammi, S.; Fontana, A.; de Laureto, P.P. Molecular insights into the interaction between alpha-synuclein and docosahexaenoic acid. J. Mol. Biol. 2009, 394, 94–107. [Google Scholar] [CrossRef]
- Broersen, K.; Ruiperez, V.; Davletov, B. Structural and Aggregation Properties of Alpha-Synuclein Linked to Phospholipase A2 Action. Protein Pept. Lett. 2018, 25, 368–378. [Google Scholar] [CrossRef]
- Iljina, M.; Tosatto, L.; Choi, M.L.; Sang, J.C.; Ye, Y.; Hughes, C.D.; Bryant, C.E.; Gandhi, S.; Klenerman, D. Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein. Sci. Rep. 2016, 6, 33928. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.H.C. Dopamine prevents lipid peroxidation-induced accumulation of toxic α-synuclein oligomers by preserving autophagy-lysosomal function. Front. Cell. Neurosci. 2013, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yamada, N.; Maruyama, W.; Osawa, T. Formation of dopamine adducts derived from brain polyunsaturated fatty acids: Mechanism for Parkinson disease. J. Biol. Chem. 2008, 283, 34887–34895. [Google Scholar] [CrossRef] [PubMed]
- Muntané, G.; Janué, A.; Fernandez, N.; Odena, M.A.; Oliveira, E.; Boluda, S.; Porero-Otin, M.; Naudí, A.; Boada, J.; Pamplona, R.; et al. Modification of brain lipids but not phenotype in alpha-synucleinopathy transgenic mice by long-term dietary n-3 fatty acids. Neurochem. Int. 2010, 56, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, K.; Kerdiles, O.; Tremblay, C.; Emond, V.; Lebel, M.; Boulianne, A.S.; Plourde, M.; Cicchetti, F.; Calon, F. Impact of DHA intake in a mouse model of synucleinopathy. Exp. Neurol. 2018, 301, 39–49. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, G.; Fecchio, C.; Sharon, R.; Schapira, A.H.V.; Proukakis, C.; Bellotti, V.; de Laureto, P.P. α-Synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J. Biol. Chem. 2017, 292, 6927–6937. [Google Scholar] [CrossRef] [PubMed]
- Fecchio, C.; Palazzi, L.; de Laureto, P.P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Swanepoel, A.C.; Buys, A.V.; Vermeulen, N.; Duim, W.; Kell, D.B. Eryptosis as a marker of Parkinson’s disease. Aging 2014, 6, 788–819. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Yu, S.Y.; Zuo, L.J.; Wang, F.; Chen, Z.J.; Hu, Y.; Wang, Y.J.; Wang, X.M.; Zhang, W. Potential biomarkers relating pathological proteins, neuroinflammatory factors and free radicals in PD patients with cognitive impairment: A cross-sectional study. BMC Neurol. 2014, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Mattammal, M.B.; Strong, R.; Lakshmi, V.M.; Chung, H.D.; Stephenson, A.H. Prostaglandin H synthetase-mediated metabolism of dopamine: Implication for Parkinson’s disease. J. Neurochem. 1995, 64, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Fang, M.; Wang, J.; Yu, H.; Hu, Z.; Yew, D.T.; Chen, W. Triptolide down-regulates COX-2 expression and PGE2 release by suppressing the activity of NF-κB and MAP kinases in lipopolysaccharide-treated PC12 cells. Phytother. Res. 2012, 26, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Zhang, T.; Fu, H.; Liu, G.X.; Wang, X.M. Schisandrin B exerts anti-neuroinflammatory activity by inhibiting the Toll-like receptor 4-dependent MyD88/IKK/NF-κB signaling pathway in lipopolysaccharide-induced microglia. Eur. J. Pharmacol. 2012, 692, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhang, X.; Li, X.; Liu, N.; Lou, F.; Ma, H.; Luo, X.; Ren, Y. Somatostatin prevents lipopolysaccharide-induced neurodegeneration in the rat substantia nigra by inhibiting the activation of microglia. Mol. Med. Rep. 2015, 12, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Song, R.; Yang, Z.; Shan, Q.; Chen, W. 6-Hydroxydopamine induces brain vascular endothelial inflammation. IUBMB Life 2017, 69, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Zhang, T.; Li, Q.; Huang, J.K.; Chen, R.F.; Sun, X.J. Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem. Int. 2013, 63, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, F.B.; Ozsoy, O.; Tanriover, G.; Kaya, Y.; Ogut, E.; Gemici, B.; Dilmac, S.; Ozkan, A.; Agar, A.; Aslan, M. Mechanism of the beneficial effect of melatonin in experimental Parkinson’s disease. Neurochem. Int. 2014, 79, 1–11. [Google Scholar] [CrossRef]
- Zhou, F.; Yao, H.H.; Wu, J.Y.; Ding, J.H.; Sun, T.; Hu, G. Opening of microglial K(ATP) channels inhibits rotenone-induced neuroinflammation. J. Cell. Mol. Med. 2008, 12, 1559–1570. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhu, X.Z. Rotenone-induced neurotoxicity of THP-1 cells requires production of reactive oxygen species and activation of phosphatidylinositol 3-kinase. Brain Res. 2007, 1153, 12–19. [Google Scholar] [CrossRef]
- Wang, T.; Pei, Z.; Zhang, W.; Liu, B.; Langenbach, R.; Lee, C.; Wilson, B.; Reece, J.M.; Miller, D.S.; Hong, J.S. MPP+-induced COX-2 activation and subsequent dopaminergic neurodegeneration. FASEB J. 2005, 19, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, O.; Tanriover, G.; Derin, N.; Uysal, N.; Demir, N.; Gemici, B.; Kencebay, C.; Yargicoglu, P.; Agar, A.; Aslan, M. The effect of docosahexaenoic Acid on visual evoked potentials in a mouse model of Parkinson’s disease: The role of cyclooxygenase-2 and nuclear factor kappa-B. Neurotox. Res. 2011, 20, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.M.; Zhang, J. Identification of proteins involved in microglial endocytosis of alpha-synuclein. J. Proteome Res. 2007, 6, 3614–3627. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Branchi, I.; D’Andrea, I.; Armida, M.; Carnevale, D.; Ajmone-Cat, M.A.; Pèzzola, A.; Potenza, R.L.; Morgese, M.G.; Cassano, T.; Minghetti, L.; et al. Striatal 6-OHDA lesion in mice: Investigating early neurochemical changes underlying Parkinson’s disease. Behav. Brain Res. 2010, 208, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef]
- Ahmad, A.S.; Maruyama, T.; Narumiya, S.; Doré, S. PGE2 EP1 receptor deletion attenuates 6-OHDA-induced Parkinsonism in mice: Old switch, new target. Neurotox. Res. 2013, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, E.; Casper, D.; Werner, P. PGE(2) receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE(2) neurotoxicity. J. Neurosci. Res. 2007, 85, 3109–3117. [Google Scholar] [CrossRef]
- Carrasco, E.; Werner, P.; Casper, D. Prostaglandin receptor EP2 protects dopaminergic neurons against 6-OHDA-mediated low oxidative stress. Neurosci. Lett. 2008, 441, 44–49. [Google Scholar] [CrossRef]
- Pradhan, S.S.; Salinas, K.; Garduno, A.C.; Johansson, J.U.; Wang, Q.; Manning-Bog, A.; Andreasson, K.I. Anti-Inflammatory and Neuroprotective Effects of PGE2 EP4 Signaling in Models of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Hinds, A.I.; Hanneman, W.H.; Tjalkens, R.B.; Legare, M.E. DJ-1 mutation decreases astroglial release of inflammatory mediators. Neurotoxicology 2016, 52, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Parga, J.A.; García-Garrote, M.; Martínez, S.; Raya, Á.; Labandeira-García, J.L.; Rodríguez-Pallares, J. Prostaglandin EP2 Receptors Mediate Mesenchymal Stromal Cell-Neuroprotective Effects on Dopaminergic Neurons. Mol. Neurobiol. 2018, 55, 4763–4776. [Google Scholar] [CrossRef]
- Wang, X.; Qin, Z.H.; Leng, Y.; Wang, Y.; Jin, X.; Chase, T.N.; Bennett, M.C. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J. Neurochem. 2002, 83, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Fukuhara, A.; Inui, T.; Allhorn, M. Prevention of paraquat-induced apoptosis in human neuronal SH-SY5Y cells by lipocalin-type prostaglandin D synthase. J. Neurochem. 2012, 120, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Weng, C.F.; Yu, N.C.; Liou, D.Y.; Kuo, F.S.; Huang, M.C.; Tam, K.; Shyue, S.K.; Cheng, H. Enhanced prostacyclin synthesis by adenoviral gene transfer reduced glial activation and ameliorated dopaminergic dysfunction in hemiparkinsonian rats. Oxid. Med. Cell. Longev. 2013, 2013, 649809. [Google Scholar] [CrossRef] [PubMed]
- Ogburn, K.D.; Figueiredo-Pereira, M.E. Cytoskeleton/endoplasmic reticulum collapse induced by prostaglandin J2 parallels centrosomal deposition of ubiquitinated protein aggregates. J. Biol. Chem. 2006, 281, 23274–23284. [Google Scholar] [CrossRef] [PubMed]
- Shivers, K.Y.; Nikolopoulou, A.; Machlovi, S.I.; Vallabhajosula, S.; Figueiredo-Pereira, M.E. PACAP27 prevents Parkinson-like neuronal loss and motor deficits but not microglia activation induced by prostaglandin J2. Biochim. Biophys. Acta 2014, 1842, 1707–1719. [Google Scholar] [CrossRef]
- Pierre, S.R.; Lemmens, M.A.M.; Figueiredo-Pereira, M.E. Subchronic infusion of the product of inflammation prostaglandin J2 models sporadic Parkinson’s disease in mice. J. Neuroinflamm. 2009, 6, 18. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Zhao, N.; Li, W.; Yang, Z.; Liu, X.; Le, W.; Zhang, X. Potential biomarkers of Parkinson’s disease revealed by plasma metabolic profiling. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1081–1082, 101–108. [Google Scholar] [CrossRef]
- Kang, K.H.; Liou, H.H.; Hour, M.J.; Liou, H.C.; Fu, W.M. Protection of dopaminergic neurons by 5-lipoxygenase inhibitor. Neuropharmacology 2013, 73, 380–387. [Google Scholar] [CrossRef]
- Nagarajan, V.B.; Marathe, P.A. Effect of montelukast in experimental model of Parkinson’s disease. Neurosci. Lett. 2018, 682, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Mansour, R.M.; Ahmed, M.A.E.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Chou, V.P.; Ko, N.; Holman, T.R.; Manning-Boğ, A.B. Gene-environment interaction models to unmask susceptibility mechanisms in Parkinson’s disease. J. Vis. Exp. 2014, e50960. [Google Scholar] [CrossRef] [PubMed]
- Searles Nielsen, S.; Bammler, T.K.; Gallagher, L.G.; Farin, F.M.; Longstreth, W.; Franklin, G.M.; Swanson, P.D.; Checkoway, H. Genotype and age at Parkinson disease diagnosis. Int. J. Mol. Epidemiol. Genet. 2013, 4, 61–69. [Google Scholar] [PubMed]
- Lakkappa, N.; Krishnamurthy, P.T.; Hammock, B.D.; Velmurugan, D.; Bharath, M.M.S. Possible role of Epoxyeicosatrienoic acid in prevention of oxidative stress mediated neuroinflammation in Parkinson disorders. Med. Hypotheses 2016, 93, 161–165. [Google Scholar] [CrossRef]
- Terashvili, M.; Sarkar, P.; Nostrand, M.V.; Falck, J.R.; Harder, D.R. The protective effect of astrocyte-derived 14, 15-epoxyeicosatrienoic acid on hydrogen peroxide-induced cell injury in astrocyte-dopaminergic neuronal cell line co-culture. Neuroscience 2012, 223, 68–76. [Google Scholar] [CrossRef]
- Qin, X.; Wu, Q.; Lin, L.; Sun, A.; Liu, S.; Li, X.; Cao, X.; Gao, T.; Luo, P.; et al. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates MPTP-Induced Parkinsonism. Mol. Neurobiol. 2015, 52, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.I.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef] [PubMed]
- Lakkappa, N.; Krishnamurthy, P.T.; Yamjala, K.; Hwang, S.H.; Hammock, B.D.; Babu, B. Evaluation of antiparkinson activity of PTUPB by measuring dopamine and its metabolites in Drosophila melanogaster: LC-MS/MS method development. J. Pharm. Biomed. Anal. 2018, 149, 457–464. [Google Scholar] [CrossRef]
- Connolly, J.; Siderowf, A.; Clark, C.M.; Mu, D.; Pratico, D. F2 isoprostane levels in plasma and urine do not support increased lipid peroxidation in cognitively impaired Parkinson disease patients. Cogn. Behav. Neurol. 2008, 21, 83–86. [Google Scholar] [CrossRef]
- Lee, C.Y.J.; Seet, R.C.S.; Huang, S.H.; Long, L.H.; Halliwell, B. Different patterns of oxidized lipid products in plasma and urine of dengue fever, stroke, and Parkinson’s disease patients: Cautions in the use of biomarkers of oxidative stress. Antioxid. Redox Signal. 2009, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.S.; Lee, C.Y.J.; Lim, E.C.H.; Tan, J.J.H.; Quek, A.M.L.; Chong, W.L.; Looi, W.F.; Huang, S.H.; Wang, H.; Chang, Y.H.; et al. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Yao, Y.; Hyman, B.T.; Growdon, J.H.; Praticò, D. Plasma F2A isoprostane levels in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2007, 4, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J.P.; Hulette, C.; Powell, S.; Roberts, L.J.; Zhang, J. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson’s disease and with dementia with Lewy body disease. J. Neurochem. 2003, 85, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Neely, M.D.; Davison, C.A.; Aschner, M.; Bowman, A.B. From the Cover: Manganese and Rotenone-Induced Oxidative Stress Signatures Differ in iPSC-Derived Human Dopamine Neurons. Toxicol. Sci. 2017, 159, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gao, X.; Yang, C.; Chen, L.; Chen, Z. Resolvin D1 Attenuates Mpp+-Induced Parkinson Disease via Inhibiting Inflammation in PC12 Cells. Med. Sci. Monit. 2017, 23, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Y.; Zhang, R.; Qiao, S.; Fan, J. Resolvin D2 recovers neural injury by suppressing inflammatory mediators expression in lipopolysaccharide-induced Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 2015, 460, 799–805. [Google Scholar] [CrossRef]
- Traina, G. The neurobiology of acetyl-l-carnitine. Front. Biosci. 2016, 21, 1314–1329. [Google Scholar] [CrossRef]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef]
- Crooks, S.A.; Bech, S.; Halling, J.; Christiansen, D.H.; Ritz, B.; Petersen, M.S. Carnitine levels and mutations in the SLC22A5 gene in Faroes patients with Parkinson’s disease. Neurosci. Lett. 2018, 675, 116–119. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Rubio, J.C.; Molina, J.A.; Martín, M.A.; Campos, Y.; Benito-León, J.; Ortí-Pareja, M.; Gassalla, T.; Arenas, J. Cerebrospinal fluid carnitine levels in patients with Parkinson’s disease. J. Neurol. Sci. 1997, 145, 183–185. [Google Scholar] [CrossRef]
- Zhang, H.; Jia, H.; Liu, J.; Ao, N.; Yan, B.; Shen, W.; Wang, X.; Li, X.; Luo, C.; Liu, J. Combined R-alpha-lipoic acid and acetyl-l-carnitine exerts efficient preventative effects in a cellular model of Parkinson’s disease. J. Cell. Mol. Med. 2010, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sadovova, N.; Ali, H.K.; Duhart, H.M.; Fu, X.; Zou, X.; Patterson, T.A.; Ninienda, Z.K.; Virmani, A.; Paule, M.G.; et al. L-carnitine protects neurons from 1-methyl-4-phenylpyridinium-induced neuronal apoptosis in rat forebrain culture. Neuroscience 2007, 144, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Gill, E.L.; Raman, S.; Yost, R.A.; Garrett, T.J.; Vedam-Mai, V. l-Carnitine Inhibits Lipopolysaccharide-Induced Nitric Oxide Production of SIM-A9 Microglia Cells. ACS Chem. Neurosci. 2018, 9, 901–905. [Google Scholar] [CrossRef]
- Singh, S.; Mishra, A.; Mishra, S.K.; Shukla, S. ALCAR promote adult hippocampal neurogenesis by regulating cell-survival and cell death-related signals in rat model of Parkinson’s disease like-phenotypes. Neurochem. Int. 2017, 108, 388–396. [Google Scholar] [CrossRef]
- Afshin-Majd, S.; Bashiri, K.; Kiasalari, Z.; Baluchnejadmojarad, T.; Sedaghat, R.; Roghani, M. Acetyl-l-carnitine protects dopaminergic nigrostriatal pathway in 6-hydroxydopamine-induced model of Parkinson’s disease in the rat. Biomed. Pharmacother. 2017, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mishra, A.; Srivastava, N.; Shukla, R.; Shukla, S. Acetyl-l-Carnitine via Upegulating Dopamine D1 Receptor and Attenuating Microglial Activation Prevents Neuronal Loss and Improves Memory Functions in Parkinsonian Rats. Mol. Neurobiol. 2018, 55, 583–602. [Google Scholar] [CrossRef]
- Sarkar, S.; Gough, B.; Raymick, J.; Beaudoin, M.A.; Ali, S.F.; Virmani, A.; Binienda, Z.K. Histopathological and electrophysiological indices of rotenone-evoked dopaminergic toxicity: Neuroprotective effects of acetyl-l-carnitine. Neurosci. Lett. 2015, 606, 53–59. [Google Scholar] [CrossRef]
- Zaitone, S.A.; Abo-Elmatty, D.M.; Shaalan, A.A. Acetyl-l-carnitine and α-lipoic acid affect rotenone-induced damage in nigral dopaminergic neurons of rat brain, implication for Parkinson’s disease therapy. Pharmacol. Biochem. Behav. 2012, 100, 347–360. [Google Scholar] [CrossRef]
- Bodis-Wollner, I.; Chung, E.; Ghilardi, M.F.; Glover, A.; Onofrj, M.; Pasik, P.; Samson, Y. Acetyl-levo-carnitine protects against MPTP-induced parkinsonism in primates. J. Neural Transm. Park Dis. Dement. Sect. 1991, 3, 63–72. [Google Scholar] [CrossRef]
- Vetel, S.; Sérrière, S.; Vercouillie, J.; Vergote, J.; Chicheri, G.; Deloye, J.B.; Dollé, F.; Bodard, S.; Tronel, C.; Nadal-Desbarats, L.; et al. Extensive exploration of a novel rat model of Parkinson’s disease using partial 6-hydroxydopamine lesion of dopaminergic neurons suggests new therapeutic approaches. Synapse 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, S.; Lu, F.; Liu, C.; Wang, Y.; Bai, Y.; Wang, N.; Liu, S.M. Cerebral metabonomics study on Parkinson’s disease mice treated with extract of Acanthopanax senticosus harms. Phytomedicine 2013, 20, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Tu-Sekine, B.; Goldschmidt, H.; Raben, D.M. Diacylglycerol, phosphatidic acid, and their metabolic enzymes in synaptic vesicle recycling. Adv. Biol. Regul. 2015, 57, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Almena, M.; Mérida, I. Shaping up the membrane: Diacylglycerol coordinates spatial orientation of signaling. Trends Biochem. Sci. 2011, 36, 593–603. [Google Scholar] [CrossRef]
- Ahmadian, M.; Duncan, R.E.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Triacylglycerol metabolism in adipose tissue. Future Lipidol. 2007, 2, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, F.; García-Gutiérrez, M.S.; Aracil-Fernández, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid CB1 and CB2 Receptors, and Monoacylglycerol Lipase Gene Expression Alterations in the Basal Ganglia of Patients with Parkinson’s Disease. Neurotherapeutics 2018, 15, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef]
- Aymerich, M.S.; Rojo-Bustamante, E.; Molina, C.; Celorrio, M.; Sánchez-Arias, J.A.; Franco, R. Neuroprotective Effect of JZL184 in MPP(+)-Treated SH-SY5Y Cells Through CB2 Receptors. Mol. Neurobiol. 2016, 53, 2312–2319. [Google Scholar] [CrossRef]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef]
- Fernández-Suárez, D.; Celorrio, M.; Riezu-Boj, J.I.; Ugarte, A.; Pacheco, R.; González, H.; Oyarzabal, J.; Hillard, C.J.; Franco, R.; Aymerich, M.S. Monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol. Aging 2014, 35, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Stampanoni Bassi, M.; Sancesario, A.; Morace, R.; Centonze, D.; Iezzi, E. Cannabinoids in Parkinson’s Disease. Cannabis Cannabinoid Res. 2017, 2, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agrò, A.; Bernardi, G.; Bursa, L.; Pierantozzi, M.; Stanzione, P.; et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann. Neurol. 2005, 57, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Pisani, V.; Moschella, V.; Bari, M.; Fezza, F.; Galati, S.; Bernardi, G.; Stacione, P.; Pisani, A.; Maccarrone, M. Dynamic changes of anandamide in the cerebrospinal fluid of Parkinson’s disease patients. Mov. Disord. 2010, 25, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Chagas, M.H.N.; Zuardi, A.W.; Tumas, V.; Pena-Pereira, M.A.; Sobreira, E.T.; Bergamaschi, M.M.; dos Santos, A.C.; Teixeira, A.L.; Hallack, J.E.; Crippa, J.A. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: An exploratory double-blind trial. J. Psychopharmacol. 2014, 28, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Oki, M.; Kaneko, S.; Morise, S.; Takenouchi, N.; Hashizume, T.; Tsuge, A.; Nakamura, M.; Wate, R.; Kausaka, H. Zonisamide ameliorates levodopa-induced dyskinesia and reduces expression of striatal genes in Parkinson model rats. Neurosci. Res. 2017, 122, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Mackovski, N.; Liao, J.; Weng, R.; Wei, X.; Wang, R.; Chen, Z.; Liu, X.; Yu, Y.; Meyer, B.J.; Xia, Y.; et al. Reversal effect of simvastatin on the decrease in cannabinoid receptor 1 density in 6-hydroxydopamine lesioned rat brains. Life Sci. 2016, 155, 123–132. [Google Scholar] [CrossRef]
- Casteels, C.; Lauwers, E.; Baitar, A.; Bormans, G.; Baekelandt, V.; Van Laere, K. In vivo type 1 cannabinoid receptor mapping in the 6-hydroxydopamine lesion rat model of Parkinson’s disease. Brain Res. 2010, 1316, 153–162. [Google Scholar] [CrossRef]
- Walsh, S.; Mnich, K.; Mackie, K.; Gorman, A.M.; Finn, D.P.; Dowd, E. Loss of cannabinoid CB1 receptor expression in the 6-hydroxydopamine-induced nigrostriatal terminal lesion model of Parkinson’s disease in the rat. Brain Res. Bull. 2010, 81, 543–548. [Google Scholar] [CrossRef]
- Chaves-Kirsten, G.P.; Mazucanti, C.H.Y.; Real, C.C.; Souza, B.M.; Britto, L.R.G.; Torrão, A.S. Temporal changes of CB1 cannabinoid receptor in the basal ganglia as a possible structure-specific plasticity process in 6-OHDA lesioned rats. PLoS ONE 2013, 8, e76874. [Google Scholar] [CrossRef]
- Silverdale, M.A.; McGuire, S.; McInnes, A.; Crossman, A.R.; Brotchie, J.M. Striatal cannabinoid CB1 receptor mRNA expression is decreased in the reserpine-treated rat model of Parkinson’s disease. Exp. Neurol. 2001, 169, 400–406. [Google Scholar] [CrossRef] [PubMed]
- González, S.; Mena, M.A.; Lastres-Becker, I.; Serrano, A.; de Yébenes, J.G.; Ramos, J.A.; Fernández-Ruiz, J. Cannabinoid CB(1) receptors in the basal ganglia and motor response to activation or blockade of these receptors in parkin-null mice. Brain Res. 2005, 1046, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Madeo, G.; Schirinzi, T.; Maltese, M.; Martella, G.; Rapino, C.; Fezza, F.; Mastrengelo, N.; Bonsi, P.; Maccarrone, M.; Pisani, A. Dopamine-dependent CB1 receptor dysfunction at corticostriatal synapses in homozygous PINK1 knockout mice. Neuropharmacology 2016, 101, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Concannon, R.M.; Okine, B.N.; Finn, D.P.; Dowd, E. Upregulation of the cannabinoid CB2 receptor in environmental and viral inflammation-driven rat models of Parkinson’s disease. Exp. Neurol. 2016, 283, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Concannon, R.M.; Okine, B.N.; Finn, D.P.; Dowd, E. Differential upregulation of the cannabinoid CB2 receptor in neurotoxic and inflammation-driven rat models of Parkinson’s disease. Exp. Neurol. 2015, 269, 133–141. [Google Scholar] [CrossRef]
- Palomo-Garo, C.; Gómez-Gálvez, Y.; García, C.; Fernández-Ruiz, J. Targeting the cannabinoid CB2 receptor to attenuate the progression of motor deficits in LRRK2-transgenic mice. Pharmacol. Res. 2016, 110, 181–192. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef]
- Fernandez-Espejo, E.; Caraballo, I.; Rodriguez de Fonseca, F.; Ferrer, B.; El Banoua, F.; Flores, J.A.; Galan-Rodriguez, B. Experimental parkinsonism alters anandamide precursor synthesis, and functional deficits are improved by AM404: A modulator of endocannabinoid function. Neuropsychopharmacology 2004, 29, 1134–1142. [Google Scholar] [CrossRef]
- Maccarrone, M.; Gubellini, P.; Bari, M.; Picconi, B.; Battista, N.; Centonze, D.; Bernardi, G.; Finazzi-Agrò, A.; Calabresi, P. Levodopa treatment reverses endocannabinoid system abnormalities in experimental parkinsonism. J. Neurochem. 2003, 85, 1018–1025. [Google Scholar] [CrossRef]
- Gubellini, P.; Picconi, B.; Bari, M.; Battista, N.; Calabresi, P.; Centonze, D.; Bernardi, G.; Finazzi-Agrò, A.; Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J. Neurosci. 2002, 22, 6900–6907. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, B.; Asbrock, N.; Kathuria, S.; Piomelli, D.; Giuffrida, A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: Implications for the treatment of levodopa-induced dyskinesias. Eur. J. Neurosci. 2003, 18, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Van der Stelt, M.; Fox, S.H.; Hill, M.; Crossman, A.R.; Petrosino, S.; Di Marzo, V.; Brotchie, J.M. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson’s disease. FASEB J. 2005, 19, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Viveros-Paredes, J.M.; Gonzalez-Castañeda, R.E.; Escalante-Castañeda, A.; Tejeda-Martínez, A.R.; Castañeda-Achutiguí, F.; Flores-Soto, M.E. Effect of inhibition of fatty acid amide hydrolase on MPTP-induced dopaminergic neuronal damage. Neurologia 2017. [Google Scholar] [CrossRef]
- Celorrio, M.; Fernández-Suárez, D.; Rojo-Bustamante, E.; Echeverry-Alzate, V.; Ramírez, M.J.; Hillard, C.J.; López-Morenoa, J.A.; Maldonado, R.; Oyarzábal, J.; Franco, R.; et al. Fatty acid amide hydrolase inhibition for the symptomatic relief of Parkinson’s disease. Brain Behav. Immun. 2016, 57, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Mnich, K.; Finn, D.P.; Dowd, E.; Gorman, A.M. Inhibition by anandamide of 6-hydroxydopamine-induced cell death in PC12 cells. Int. J. Cell Biol. 2010, 2010, 818497. [Google Scholar] [CrossRef]
- Mounsey, R.B.; Mustafa, S.; Robinson, L.; Ross, R.A.; Riedel, G.; Pertwee, R.G.; Tesimann, P. Increasing levels of the endocannabinoid 2-AG is neuroprotective in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson’s disease. Exp. Neurol. 2015, 273, 36–44. [Google Scholar] [CrossRef]
- Hracskó, Z.; Baranyi, M.; Csölle, C.; Gölöncsér, F.; Madarász, E.; Kittel, A.; Sperlágh, B. Lack of neuroprotection in the absence of P2X7 receptors in toxin-induced animal models of Parkinson’s disease. Mol. Neurodegener. 2011, 6, 28. [Google Scholar] [CrossRef]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar]
- Freestone, P.S.; Guatteo, E.; Piscitelli, F.; di Marzo, V.; Lipski, J.; Mercuri, N.B. Glutamate spillover drives endocannabinoid production and inhibits GABAergic transmission in the Substantia Nigra pars compacta. Neuropharmacology 2014, 79, 467–475. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Malenka, R.C. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 2007, 445, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; et al. Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: A lipidomic analysis. PLoS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M.; et al. Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef] [PubMed]
- Simón-Sánchez, J.; van Hilten, J.J.; van de Warrenburg, B.; Post, B.; Berendse, H.W.; Arepalli, S.; Hernandez, D.G.; de Bie, R.M.; Velseboar, D.; Scheffer, H.; et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur. J. Hum. Genet. 2011, 19, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Song, W.; Huang, R.; Chen, K.; Zhao, B.; Li, J.; Yang, Y.; Shang, H.F. GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population. J. Clin. Neurosci. 2013, 20, 880–883. [Google Scholar] [CrossRef]
- Sakane, F.; Mizuno, S.; Takahashi, D.; Sakai, H. Where do substrates of diacylglycerol kinases come from? Diacylglycerol kinases utilize diacylglycerol species supplied from phosphatidylinositol turnover-independent pathways. Adv. Biol. Regul. 2018, 67, 101–108. [Google Scholar] [CrossRef]
- Guo, X.; Song, W.; Chen, K.; Chen, X.; Zheng, Z.; Cao, B.; Huang, R.; Zhao, B.; Wu, Y.; Shang, H.F. The serum lipid profile of Parkinson’s disease patients: A study from China. Int. J. Neurosci. 2015, 125, 838–844. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, H.; Tian, Y.; Xu, F.; Chen, X.; Wang, K. Reduced serum levels of triglyceride, very low density lipoprotein cholesterol and apolipoprotein B in Parkinson’s disease patients. PLoS ONE 2013, 8, e75743. [Google Scholar] [CrossRef] [PubMed]
- Gregório, M.L.; Pinhel, M.A.S.; Sado, C.L.; Longo, G.S.; Oliveira, F.N.; Amorim, G.S.; Nakazone, M.A.; Florim, G.M.; Mazeti, C.M.; Martins, D.P.; et al. Impact of genetic variants of apolipoprotein E on lipid profile in patients with Parkinson’s disease. Biomed. Res. Int. 2013, 2013, 641515. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Cassani, E.; Barichella, M.; Spadafranca, A.; Caccialanza, R.; Bertoli, S.; Battezzati, A.; Pezzoli, G. Low cardiometabolic risk in Parkinson’s disease is independent of nutritional status, body composition and fat distribution. Clin. Nutr. 2012, 31, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Sääksjärvi, K.; Knekt, P.; Männistö, S.; Lyytinen, J.; Heliövaara, M. Prospective study on the components of metabolic syndrome and the incidence of Parkinson’s disease. Park. Relat. Disord. 2015, 21, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Vikdahl, M.; Bäckman, L.; Johansson, I.; Forsgren, L.; Håglin, L. Cardiovascular risk factors and the risk of Parkinson’s disease. Eur. J. Clin. Nutr. 2015, 69, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Scigliano, G.; Musicco, M.; Soliveri, P.; Piccolo, I.; Ronchetti, G.; Girotti, F. Reduced risk factors for vascular disorders in Parkinson disease patients: A case-control study. Stroke 2006, 37, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Hishikawa, N.; Shang, J.; Sato, K.; Nakano, Y.; Morihara, R.; Ohta, Y.; Yamashita, T.; Abe, K. Peripheral arterial endothelial dysfunction of neurodegenerative diseases. J. Neurol. Sci. 2016, 366, 94–99. [Google Scholar] [CrossRef]
- Ya, L.; Lu, Z. Differences in ABCA1 R219K Polymorphisms and Serum Indexes in Alzheimer and Parkinson Diseases in Northern China. Med. Sci. Monit. 2017, 23, 4591–4600. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Guerreiro, P.S.; Coelho, J.E.; Sousa-Lima, I.; Macedo, P.; Lopes, L.V.; Outeiro, T.F.; Pais, T.F. Mutant A53T α-Synuclein Improves Rotarod Performance Before Motor Deficits and Affects Metabolic Pathways. Neuromol. Med. 2017, 19, 113–121. [Google Scholar] [CrossRef]
- Meng, X.; Zheng, R.; Zhang, Y.; Qiao, M.; Liu, L.; Jing, P.; Wang, L.; Liu, J.; Gao, Y. An activated sympathetic nervous system affects white adipocyte differentiation and lipolysis in a rat model of Parkinson’s disease. J. Neurosci. Res. 2015, 93, 350–360. [Google Scholar] [CrossRef]
- He, Q.; Wang, M.; Petucci, C.; Gardell, S.J.; Han, X. Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. Int. J. Biochem. Cell Biol. 2013, 45, 2749–2755. [Google Scholar] [CrossRef]
- Sere, Y.Y.; Regnacq, M.; Colas, J.; Berges, T. A Saccharomyces cerevisiae strain unable to store neutral lipids is tolerant to oxidative stress induced by α-synuclein. Free Radic. Biol. Med. 2010, 49, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef]
- Sánchez Campos, S.; Alza, N.P.; Salvador, G.A. Lipid metabolism alterations in the neuronal response to A53T α-synuclein and Fe-induced injury. Arch. Biochem. Biophys. 2018, 655, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Antonny, B.; Vanni, S.; Shindou, H.; Ferreira, T. From zero to six double bonds: Phospholipid unsaturation and organelle function. Trends Cell Biol. 2015, 25, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Grinstein, S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol. Rev. 2013, 93, 69–106. [Google Scholar] [CrossRef]
- Zhang, Q.; Tamura, Y.; Roy, M.; Adachi, Y.; Iijima, M.; Sesaki, H. Biosynthesis and roles of phospholipids in mitochondrial fusion, division and mitophagy. Cell. Mol. Life Sci. 2014, 71, 3767–3778. [Google Scholar] [CrossRef]
- Boss, W.F.; Im, Y.J. Phosphoinositide signaling. Annu. Rev. Plant Biol. 2012, 63, 409–429. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid signaling in the nervous system. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Phosphatidylserine-mediated cellular signaling. Adv. Exp. Med. Biol. 2013, 991, 177–193. [Google Scholar] [CrossRef]
- Musille, P.M.; Kohn, J.A.; Ortlund, E.A. Phospholipid—Driven gene regulation. FEBS Lett. 2013, 587, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, Y.; Wang, X. Phosphatidic acid-mediated signaling. Adv. Exp. Med. Biol. 2013, 991, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Ammar, M.R.; Kassas, N.; Bader, M.F.; Vitale, N. Phosphatidic acid in neuronal development: A node for membrane and cytoskeleton rearrangements. Biochimie 2014, 107, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Frohman, M.A. Mitochondria: Signaling with phosphatidic acid. Int. J. Biochem. Cell Biol. 2012, 44, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Hess, S.K.; Heinrich, F.; Lee, J.C. Molecular details of α-synuclein membrane association revealed by neutrons and photons. J. Phys. Chem. B 2015, 119, 4812–4823. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Sasai, H.; Kume, A.; Takahashi, D.; Satoh, M.; Kado, S.; Sakane, F. Dioleoyl-phosphatidic acid selectively binds to α-synuclein and strongly induces its aggregation. FEBS Lett. 2017, 591, 784–791. [Google Scholar] [CrossRef]
- Soper, J.H.; Kehm, V.; Burd, C.G.; Bankaitis, V.A.; Lee, V.M.Y. Aggregation of α-synuclein in S. cerevisiae is associated with defects in endosomal trafficking and phospholipid biosynthesis. J. Mol. Neurosci. 2011, 43, 391–405. [Google Scholar] [CrossRef]
- Holemans, T.; Sørensen, D.M.; van Veen, S.; Martin, S.; Hermans, D.; Kemmer, G.C.; Van den Haute, C.; Baekelandt, V.; Günther Pomorski, T.; Agostinis, P.; et al. A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc. Natl. Acad. Sci. USA 2015, 112, 9040–9045. [Google Scholar] [CrossRef]
- Martin, S.; van Veen, S.; Holemans, T.; Demirsoy, S.; van den Haute, C.; Baekelandt, V.; Agostinis, P.; Eggermont, J.; Vangheluwe, P. Protection against Mitochondrial and Metal Toxicity Depends on Functional Lipid Binding Sites in ATP13A2. Park. Dis. 2016, 2016, 9531917. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Gomez, H.R.; Singh, J.; Meyers, C.; Chen, W.; Gorbatyuk, O.S.; Muzyczka, N. The Lipase Activity of Phospholipase D2 is Responsible for Nigral Neurodegeneration in a Rat Model of Parkinson’s Disease. Neuroscience 2018, 377, 174–183. [Google Scholar] [CrossRef]
- Binder, B.Y.K.; Williams, P.A.; Silva, E.A.; Leach, J.K. Lysophosphatidic Acid and Sphingosine-1-Phosphate: A Concise Review of Biological Function and Applications for Tissue Engineering. Tissue Eng. Part B Rev. 2015, 21, 531–542. [Google Scholar] [CrossRef]
- Sheng, X.; Yung, Y.C.; Chen, A.; Chun, J. Lysophosphatidic acid signalling in development. Development 2015, 142, 1390–1395. [Google Scholar] [CrossRef]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef]
- Yang, X.Y.; Zhao, E.Y.; Zhuang, W.X.; Sun, F.X.; Han, H.L.; Han, H.R.; Lin, Z.J.; Pan, Z.F.; Qu, M.H.; Zeng, X.W.; et al. LPA signaling is required for dopaminergic neuron development and is reduced through low expression of the LPA1 receptor in a 6-OHDA lesion model of Parkinson’s disease. Neurol. Sci. 2015, 36, 2027–2033. [Google Scholar] [CrossRef]
- Choi, J.H.; Jang, M.; Oh, S.; Nah, S.Y.; Cho, I.H. Multi-Target Protective Effects of Gintonin in 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-Mediated Model of Parkinson’s Disease via Lysophosphatidic Acid Receptors. Front. Pharmacol. 2018, 9, 515. [Google Scholar] [CrossRef]
- Vance, J.E. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: Two metabolically related aminophospholipids. J. Lipid Res. 2008, 49, 1377–1387. [Google Scholar] [CrossRef]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alacaly, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Park. Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef]
- Riekkinen, P.; Rinne, U.K.; Pelliniemi, T.T.; Sonninen, V. Interaction between dopamine and phospholipids. Studies of the substantia nigra in Parkinson disease patients. Arch. Neurol. 1975, 32, 25–27. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10. [Google Scholar] [CrossRef]
- Ross, B.M.; Mamalias, N.; Moszczynska, A.; Rajput, A.H.; Kish, S.J. Elevated activity of phospholipid biosynthetic enzymes in substantia nigra of patients with Parkinson’s disease. Neuroscience 2001, 102, 899–904. [Google Scholar] [CrossRef]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, S.D.; Hulleman, J.D.; Dutseva, E.A.; Antonenko, Y.N.; Rochet, J.C.; Cramer, W.A. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry 2007, 46, 14369–14379. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, S.; Liou, L.C.; Ren, Q.; Zhang, Z.; Caldwell, G.A.; Witt, S.N. Phosphatidylethanolamine deficiency disrupts α-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3976–E3985. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, S.; Xu, C.; Barron, A.; Galiano, F.; Patel, D.; Lee, Y.J.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Chemical Compensation of Mitochondrial Phospholipid Depletion in Yeast and Animal Models of Parkinson’s Disease. PLoS ONE 2016, 11, e0164465. [Google Scholar] [CrossRef]
- Lee, E.S.; Charlton, C.G. 1-Methyl-4-phenyl-pyridinium increases S-adenosyl-L-methionine dependent phospholipid methylation. Pharmacol. Biochem. Behav. 2001, 70, 105–114. [Google Scholar] [CrossRef]
- Lee, E.S.Y.; Chen, H.; Charlton, C.G.; Soliman, K.F.A. The role of phospholipid methylation in 1-methyl-4-phenyl-pyridinium ion (MPP+)-induced neurotoxicity in PC12 cells. Neurotoxicology 2005, 26, 945–957. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef]
- Kim, H.Y.; Huang, B.X.; Spector, A.A. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 2014, 56, 1–18. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef]
- Valadas, J.S.; Esposito, G.; Vandekerkhove, D.; Miskiewicz, K.; Deaulmerie, L.; Raitano, S.; Seibler, P.; Lein, C.; Verstreken, P. ER Lipid Defects in Neuropeptidergic Neurons Impair Sleep Patterns in Parkinson’s Disease. Neuron 2018, 98, 1155–1169.e6. [Google Scholar] [CrossRef]
- Wei, L.; Sun, C.; Lei, M.; Li, G.; Yi, L.; Luo, F.; Li, Y.; Ding, L.; Liu, Z.; Li, S.; et al. Activation of Wnt/β-catenin pathway by exogenous Wnt1 protects SH-SY5Y cells against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013, 49, 105–115. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of apoptotic erythrocyte death by rotenone. Toxicology 2012, 300, 132–137. [Google Scholar] [CrossRef]
- González-Polo, R.A.; Niso-Santano, M.; Ortíz-Ortíz, M.A.; Gómez-Martín, A.; Morán, J.M.; García-Rubio, L.; Francisco-Mocillo, J.; Zaragoza, C.; Soler, G.; Fuentes, J.M. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol. Sci. 2007, 97, 448–458. [Google Scholar] [CrossRef]
- Ye, S.; Koon, H.K.; Fan, W.; Xu, Y.; Wei, W.; Xu, C.; Cai, J. Effect of a Traditional Chinese Herbal Medicine Formulation on Cell Survival and Apoptosis of MPP+-Treated MES 23.5 Dopaminergic Cells. Park. Dis. 2017, 2017, 4764212. [Google Scholar] [CrossRef]
- Flower, T.R.; Chesnokova, L.S.; Froelich, C.A.; Dixon, C.; Witt, S.N. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J. Mol. Biol. 2005, 351, 1081–1100. [Google Scholar] [CrossRef]
- Emmrich, J.V.; Hornik, T.C.; Neher, J.J.; Brown, G.C. Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 2013, 280, 5030–5038. [Google Scholar] [CrossRef]
- Almandoz-Gil, L.; Lindström, V.; Sigvardson, J.; Kahle, P.J.; Lannfelt, L.; Ingelsson, M.; Bergström, J. Mapping of Surface-Exposed Epitopes of In Vitro and In Vivo Aggregated Species of Alpha-Synuclein. Cell. Mol. Neurobiol. 2017, 37, 1217–1226. [Google Scholar] [CrossRef]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727. [Google Scholar] [CrossRef]
- Araki, K.; Sugawara, K.; Hayakawa, E.H.; Ubukawa, K.; Kobayashi, I.; Wakui, H.; Takahasi, N.; Sawada, K.; Mochizuki, H.; Nunomura, W. The localization of α-synuclein in the process of differentiation of human erythroid cells. Int. J. Hematol. 2018, 108, 130–138. [Google Scholar] [CrossRef]
- Hu, R.; Diao, J.; Li, J.; Tang, Z.; Li, X.; Leitz, J.; Long, J.; Liu, J.; Yu, D.; Zhao, Q. Intrinsic and membrane-facilitated α-synuclein oligomerization revealed by label-free detection through solid-state nanopores. Sci. Rep. 2016, 6, 20776. [Google Scholar] [CrossRef]
- Lou, X.; Kim, J.; Hawk, B.J.; Shin, Y.K. α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 2017, 474, 2039–2049. [Google Scholar] [CrossRef]
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef]
- Treede, I.; Braun, A.; Sparla, R.; Kühnel, M.; Giese, T.; Turner, J.R.; Anes, E.; Kulaksiz, H.; Füllekrug, J.; Stremmel, W.; et al. Anti-inflammatory effects of phosphatidylcholine. J. Biol. Chem. 2007, 282, 27155–27164. [Google Scholar] [CrossRef]
- Lagace, T.A. Phosphatidylcholine: Greasing the Cholesterol Transport Machinery. Lipid Insights 2015, 8 (Suppl. 1), 65–73. [Google Scholar] [CrossRef]
- Marcucci, H.; Paoletti, L.; Jackowski, S.; Banchio, C. Phosphatidylcholine biosynthesis during neuronal differentiation and its role in cell fate determination. J. Biol. Chem. 2010, 285, 25382–25393. [Google Scholar] [CrossRef]
- Li, T.; Tang, W.; Zhang, L. Monte Carlo cross-validation analysis screens pathway cross-talk associated with Parkinson’s disease. Neurol. Sci. 2016, 37, 1327–1333. [Google Scholar] [CrossRef]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson’s Disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef]
- Stöckl, M.; Fischer, P.; Wanker, E.; Herrmann, A. Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol. 2008, 375, 1394–1404. [Google Scholar] [CrossRef]
- Jiang, Z.; de Messieres, M.; Lee, J.C. Membrane remodeling by α-synuclein and effects on amyloid formation. J. Am. Chem. Soc. 2013, 135, 15970–15973. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Fantini, J.; Chahinian, H.; Maresca, M.; Taïeb, N.; Yahi, N. Altered ion channel formation by the Parkinson’s-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J. Mol. Biol. 2010, 397, 202–218. [Google Scholar] [CrossRef]
- O’Leary, E.I.; Jiang, Z.; Strub, M.P.; Lee, J.C. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-synuclein. J. Biol. Chem. 2018, 293, 11195–11205. [Google Scholar] [CrossRef]
- Hollie, N.I.; Cash, J.G.; Matlib, M.A.; Wortman, M.; Basford, J.E.; Abplanalp, W.; Hui, D.Y. Micromolar changes in lysophosphatidylcholine concentration cause minor effects on mitochondrial permeability but major alterations in function. Biochim. Biophys. Acta 2014, 1841, 888–895. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Li, Y.; Kuo, Y.M.; Andrews, A.J.; Nanayakkara, G.; Johnson, C.; Fu, H.; Shan, H.; Du, F.; et al. Mitochondrial Reactive Oxygen Species Mediate Lysophosphatidylcholine-Induced Endothelial Cell Activation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1090–1100. [Google Scholar] [CrossRef]
- Hung, N.D.; Sok, D.E.; Kim, M.R. Prevention of 1-palmitoyl lysophosphatidylcholine-induced inflammation by polyunsaturated acyl lysophosphatidylcholine. Inflamm. Res. 2012, 61, 473–483. [Google Scholar] [CrossRef]
- Lee, E.S.Y.; Chen, H.; Shepherd, K.R.; Lamango, N.S.; Soliman, K.F.A.; Charlton, C.G. Inhibitory effects of lysophosphatidylcholine on the dopaminergic system. Neurochem. Res. 2004, 29, 1333–1342. [Google Scholar] [CrossRef]
- Lee, E.S.Y.; Soliman, K.F.A.; Charlton, C.G. Lysophosphatidylcholine decreases locomotor activities and dopamine turnover rate in rats. Neurotoxicology 2005, 26, 27–38. [Google Scholar] [CrossRef]
- Pacheco, M.A.; Jope, R.S. Phosphoinositide signaling in human brain. Prog. Neurobiol. 1996, 50, 255–273. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Snoek, G.T.; Adamczyk, A.; Małecki, A.; Strosznajder, J.B. Phosphatidylinositol transfer protein expression altered by aging and Parkinson disease. Cell. Mol. Neurobiol. 2006, 26, 1153–1166. [Google Scholar] [CrossRef]
- Cockcroft, S. The diverse functions of phosphatidylinositol transfer proteins. Curr. Top. Microbiol. Immunol. 2012, 362, 185–208. [Google Scholar] [CrossRef]
- Lee, E.N.; Lee, S.Y.; Lee, D.; Kim, J.; Paik, S.R. Lipid interaction of alpha-synuclein during the metal-catalyzed oxidation in the presence of Cu2+ and H2O2. J. Neurochem. 2003, 84, 1128–1142. [Google Scholar] [CrossRef]
- Narayanan, V.; Guo, Y.; Scarlata, S. Fluorescence studies suggest a role for alpha-synuclein in the phosphatidylinositol lipid signaling pathway. Biochemistry 2005, 44, 462–470. [Google Scholar] [CrossRef]
- Sekar, S.; Taghibiglou, C. Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci. Lett. 2018, 666, 139–143. [Google Scholar] [CrossRef]
- Demirsoy, S.; Martin, S.; Motamedi, S.; van Veen, S.; Holemans, T.; Van den Haute, C.; Jordanova, A.; Baekelandt, V.; Vangheluwe, P.; Agostinis, P. ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum. Mol. Genet. 2017, 26, 1656–1669. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Jensen, P.H.; Marsh, D. Association of alpha-synuclein and mutants with lipid membranes: Spin-label ESR and polarized IR. Biochemistry 2006, 45, 3386–3395. [Google Scholar] [CrossRef]
- Jiang, Z.; Flynn, J.D.; Teague, W.E.; Gawrisch, K.; Lee, J.C. Stimulation of α-synuclein amyloid formation by phosphatidylglycerol micellar tubules. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef]
- Bédard, L.; Lefèvre, T.; Morin-Michaud, É.; Auger, M. Besides fibrillization: Putative role of the peptide fragment 71-82 on the structural and assembly behavior of α-synuclein. Biochemistry 2014, 53, 6463–6472. [Google Scholar] [CrossRef]
- Pandey, A.P.; Haque, F.; Rochet, J.C.; Hovis, J.S. Clustering of alpha-synuclein on supported lipid bilayers: Role of anionic lipid, protein, and divalent ion concentration. Biophys. J. 2009, 96, 540–551. [Google Scholar] [CrossRef]
- Stefanovic, A.N.D.; Claessens, M.M.A.E.; Blum, C.; Subramaniam, V. Alpha-synuclein amyloid oligomers act as multivalent nanoparticles to cause hemifusion in negatively charged vesicles. Small 2015, 11, 2257–2262. [Google Scholar] [CrossRef]
- Van Rooijen, B.D.; Claessens, M.M.A.E.; Subramaniam, V. Lipid bilayer disruption by oligomeric alpha-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta 2009, 1788, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, B.D.; Claessens, M.M.A.E.; Subramaniam, V. Membrane Permeabilization by Oligomeric α-Synuclein: In Search of the Mechanism. PLoS ONE 2010, 5, e14292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Qin, Z.J.; Hu, D.; Munishkina, L.A.; Fink, A.L. Alpha-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry 2006, 45, 8135–8142. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Phoon, C.K.L.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox. Signal. 2014, 20, 1925–1953. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Vos, M.; Geens, A.; Böhm, C.; Deaulmerie, L.; Swerts, J.; Rossi, M.; Craessaerts, K.; Leites, E.P.; Seibler, P.; Rakovic, A.; et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 2017, 216, 695–708. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Winnica, D.E.; Kapralova, V.I.; Kapralov, A.A.; Tyurin, V.A.; Kagan, V.E. LC/MS characterization of rotenone induced cardiolipin oxidation in human lymphocytes: Implications for mitochondrial dysfunction associated with Parkinson’s disease. Mol. Nutr. Food Res. 2013, 57, 1410–1422. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Polimova, A.M.; Maciel, E.; Tyurin, V.A.; Kapralova, V.I.; Winnica, D.E.; Vikulina, A.S.; Domigues, M.R.; McCoy, J.; Samders, L.H.; et al. LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: Implication for mitochondrial dysfunction in Parkinson’s disease. Free Radic. Res. 2015, 49, 681–691. [Google Scholar] [CrossRef]
- Zigoneanu, I.G.; Yang, Y.J.; Krois, A.S.; Haque, E.; Pielak, G.J. Interaction of α-synuclein with vesicles that mimic mitochondrial membranes. Biochim. Biophys. Acta 2012, 1818, 512–519. [Google Scholar] [CrossRef]
- Robotta, M.; Gerding, H.R.; Vogel, A.; Hauser, K.; Schildknecht, S.; Karreman, C.; Leist, M.; Subramaniam, V.; Drescher, M. Alpha-synuclein binds to the inner membrane of mitochondria in an α-helical conformation. Chembiochem 2014, 15, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munushkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. α-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res. 2014, 1591, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Wang, Z.; Lu, L.; Duan, C.; Wang, X.; Yang, H. Morphological analysis of mitochondria for evaluating the toxicity of α-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed. Pharmacother. 2017, 96, 1380–1388. [Google Scholar] [CrossRef]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: Protection against apoptosis versus delayed oxidative stress in Parkinson disease. J. Biol. Chem. 2009, 284, 15951–15969. [Google Scholar] [CrossRef]
- Ghio, S.; Kamp, F.; Cauchi, R.; Giese, A.; Vassallo, N. Interaction of α-synuclein with biomembranes in Parkinson’s disease—Role of cardiolipin. Prog. Lipid Res. 2016, 61, 73–82. [Google Scholar] [CrossRef]
- Chu, C.T.; Bayır, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Xu, Y.H.; Barnes, S.; Sun, Y.; Grabowski, G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010, 51, 1643–1675. [Google Scholar] [CrossRef]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef]
- Martin, R.; Sospedra, M. Sphingosine-1 phosphate and central nervous system. Curr. Top. Microbiol. Immunol. 2014, 378, 149–170. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Motyl, J.; Wencel, P.L.; Cieślik, M.; Strosznajder, R.P.; Strosznajder, J.B. Alpha-synuclein alters differently gene expression of Sirts, PARPs and other stress response proteins: Implications for neurodegenerative disorders. Mol. Neurobiol. 2018, 55, 727–740. [Google Scholar] [CrossRef]
- Zhang, L.; Okada, T.; Badawy, S.M.M.; Hirai, C.; Kajimoto, T.; Nakamura, S.I. Extracellular α-synuclein induces sphingosine 1-phosphate receptor subtype 1 uncoupled from inhibitory G-protein leaving β-arrestin signal intact. Sci. Rep. 2017, 7, 44248. [Google Scholar] [CrossRef] [PubMed]
- Badawy, S.M.M.; Okada, T.; Kajimoto, T.; Hirase, M.; Matovelo, S.A.; Nakamura, S.; Yoshida, D.; Ijuin, T.; Nakamura, S.I. Extracellular α-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J. Biol. Chem. 2018, 293, 8208–8216. [Google Scholar] [CrossRef]
- Sivasubramanian, M.; Kanagaraj, N.; Dheen, S.T.; Tay, S.S.W. Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson’s disease and in MPP+-treated MN9D cells in vitro. Neuroscience 2015, 290, 636–648. [Google Scholar] [CrossRef]
- Pyszko, J.A.; Strosznajder, J.B. The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 2014, 52, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Yang, X.; Yang, L.; Li, M.; Wood, K.; Liu, Q.; Zhu, X. Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: A pilot study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137 Pt 3, 834–848. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef]
- Halliday, G.M.; McCann, H. The progression of pathology in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 188–195. [Google Scholar] [CrossRef]
- Lin, G.; Lee, P.T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain. Cell Metab. 2018. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef]
- Arboleda, G.; Cárdenas, Y.; Rodríguez, Y.; Morales, L.C.; Matheus, L.; Arboleda, H. Differential regulation of AKT, MAPK and GSK3β during C2-ceramide-induced neuronal death. Neurotoxicology 2010, 31, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.N.; Chen, X.; Bandyopadhyay, S.; Merrill, A.H.; Tansey, M.G. Ceramide sphingolipid signaling mediates Tumor Necrosis Factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol. Neurodegener. 2012, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- France-Lanord, V.; Brugg, B.; Michel, P.P.; Agid, Y.; Ruberg, M. Mitochondrial free radical signal in ceramide-dependent apoptosis: A putative mechanism for neuronal death in Parkinson’s disease. J. Neurochem. 1997, 69, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, G.; Waters, C.; Gibson, R. Inhibition of caspases but not of calpains temporarily protect against C2-ceramide-induced death of CAD cells. Neurosci. Lett. 2007, 421, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, C.A.; Ancolio, K.; Checler, F. Wild-type but not Parkinson’s disease-related ala-53→Thr mutant alpha -synuclein protects neuronal cells from apoptotic stimuli. J. Biol. Chem. 2000, 275, 24065–24069. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Mora, R.M.; Arboleda, H.; Arboleda, G. PINK1 overexpression protects against C2-ceramide-induced CAD cell death through the PI3K/AKT pathway. J. Mol. Neurosci. 2012, 47, 582–594. [Google Scholar] [CrossRef]
- Rojas-Charry, L.; Cookson, M.R.; Niño, A.; Arboleda, H.; Arboleda, G. Downregulation of Pink1 influences mitochondrial fusion-fission machinery and sensitizes to neurotoxins in dopaminergic cells. Neurotoxicology 2014, 44, 140–148. [Google Scholar] [CrossRef]
- Jaramillo-Gómez, J.; Niño, A.; Arboleda, H.; Arboleda, G. Overexpression of DJ-1 protects against C2-ceramide-induced neuronal death through activation of the PI3K/AKT pathway and inhibition of autophagy. Neurosci. Lett. 2015, 603, 71–76. [Google Scholar] [CrossRef]
- Jung, J.S.; Shin, K.O.; Lee, Y.M.; Shin, J.A.; Park, E.M.; Jeong, J.; Kim, D.H.; Choi, J.W.; Kim, H.S. Anti-inflammatory mechanism of exogenous C2 ceramide in lipopolysaccharide-stimulated microglia. Biochim. Biophys. Acta 2013, 1831, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, X.; Duan, C.; Lu, L.; Yang, H. Alpha-synuclein overexpression increases phospho-protein phosphatase 2A levels via formation of calmodulin/Src complex. Neurochem. Int. 2013, 63, 180–194. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The novel mechanism of rotenone-induced α-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int. J. Biochem. Cell Biol. 2016, 75, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F. Sphingolipids in inflammation: Pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009, 158, 982–993. [Google Scholar] [CrossRef]
- Norris, G.H.; Blesso, C.N. Dietary and Endogenous Sphingolipid Metabolism in Chronic Inflammation. Nutrients 2017, 9, 1180. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Tommasino, C.; Marconi, M.; Ciarlo, L.; Matarrese, P.; Malorni, W. Autophagic flux and autophagosome morphogenesis require the participation of sphingolipids. Apoptosis 2015, 20, 645–657. [Google Scholar] [CrossRef]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.-Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13-5. [Google Scholar] [CrossRef]
- Mao, C.Y.; Yang, J.; Wang, H.; Zhang, S.Y.; Yang, Z.H.; Luo, H.Y.; Li, F.; Shi, M.; Liu, Y.T.; Zhuang, Z.P.; et al. SMPD1 variants in Chinese Han patients with sporadic Parkinson’s disease. Park. Relat. Disord. 2017, 34, 59–61. [Google Scholar] [CrossRef]
- Kim, W.S.; Halliday, G.M. Changes in sphingomyelin level affect alpha-synuclein and ABCA5 expression. J. Park. Dis. 2012, 2, 41–46. [Google Scholar] [CrossRef]
- Den Jager, W.A. Sphingomyelin in Lewy inclusion bodies in Parkinson’s disease. Arch. Neurol. 1969, 21, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H.V. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Kurup, R.K.; Kurup, P.A. Hypothalamic digoxin-mediated model for Parkinson’s disease. Int. J. Neurosci. 2003, 113, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Sun, Y.; Shacka, J.J.; Auray-Blais, C. Tandem Mass Spectrometry Multiplex Analysis of Glucosylceramide and Galactosylceramide Isoforms in Brain Tissues at Different Stages of Parkinson Disease. Anal. Chem. 2016, 88, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018. [Google Scholar] [CrossRef]
- Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s disease: The potential contribution of galactosylceramidase deficiency to neuronal vulnerability in late-onset synucleinopathies. J. Neurosci. Res. 2016, 94, 1328–1332. [Google Scholar] [CrossRef]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2017, 97, 92–107.e10. [Google Scholar] [CrossRef]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Fujikake, N.; Takeuchi, T.; Kohyama-Koganeya, A.; Nakajima, K.; Hirabayashi, Y.; Wada, K.; Nagai, Y. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 6675–6686. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Noelker, C.; Lu, L.; Höllerhage, M.; Vulinovic, F.; Sturn, A.; Roscher, R.; Höglinger, G.U.; Hirsch, E.C.; Oertel, W.H.; Alvarez-Fisher, D.; et al. Glucocerebrosidase deficiency and mitochondrial impairment in experimental Parkinson disease. J. Neurol. Sci. 2015, 356, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef]
- Palmano, K.; Rowan, A.; Guillermo, R.; Guan, J.; McJarrow, P. The role of gangliosides in neurodevelopment. Nutrients 2015, 7, 3891–3913. [Google Scholar] [CrossRef]
- Schnaar, R.L. Gangliosides of the Vertebrate Nervous System. J. Mol. Biol. 2016, 428, 3325–3336. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef]
- Schneider, J.S. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS ONE 2018, 13, e0199189. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Cambi, F.; Gollomp, S.M.; Kuwabara, H.; Brašić, J.R.; Leiby, B.; Sendek, S.; Wong, D.F. GM1 ganglioside in Parkinson’s disease: Pilot study of effects on dopamine transporter binding. J. Neurol. Sci. 2015, 356, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010, 292, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S. GM1 ganglioside in the treatment of Parkinson’s disease. Ann. N. Y. Acad. Sci. 1998, 845, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Roeltgen, D.P.; Mancall, E.L.; Chapas-Crilly, J.; Rothblat, D.S.; Tatarian, G.T. Parkinson’s disease: Improved function with GM1 ganglioside treatment in a randomized placebo-controlled study. Neurology 1998, 50, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Roeltgen, D.P.; Rothblat, D.S.; Chapas-Crilly, J.; Seraydarian, L.; Rao, J. GM1 ganglioside treatment of Parkinson’s disease: An open pilot study of safety and efficacy. Neurology 1995, 45, 1149–1154. [Google Scholar] [CrossRef]
- Schneider, J.S.; Seyfried, T.N.; Choi, H.S.; Kidd, S.K. Intraventricular Sialidase Administration Enhances GM1 Ganglioside Expression and Is Partially Neuroprotective in a Mouse Model of Parkinson’s Disease. PLoS ONE 2015, 10, e0143351. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, Y.; Fang, X.; Lu, Y.; Li, J.; Zhang, J.; Deng, X.; Li, S. The possible mechanism of Parkinson’s disease progressive damage and the preventive effect of GM1 in the rat model induced by 6-hydroxydopamine. Brain Res. 2014, 1592, 73–81. [Google Scholar] [CrossRef]
- Goettl, V.M.; Wemlinger, T.A.; Duchemin, A.M.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside restores dopaminergic neurochemical and morphological markers in aged rats. Neuroscience 1999, 92, 991–1000. [Google Scholar] [CrossRef]
- Emborg, M.E.; Colombo, J.A. Long-term MPTP-treated monkeys are resistant to GM1 systemic therapy. Mol. Chem. Neuropathol. 1994, 21, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Pope-Coleman, A.; Schneider, J.S. Effects of Chronic GM1 Ganglioside Treatment on Cognitieve and Motor Deficits in a Slowly Progressing Model of Parkinsonism in Non-Human Primates. Restor. Neurol. Neurosci. 1998, 12, 255–266. [Google Scholar] [PubMed]
- Herrero, M.T.; Perez-Otaño, I.; Oset, C.; Kastner, A.; Hirsch, E.C.; Agid, Y.; Luguin, M.R.; Obeso, J.A.; Del Río, J. GM-1 ganglioside promotes the recovery of surviving midbrain dopaminergic neurons in MPTP-treated monkeys. Neuroscience 1993, 56, 965–972. [Google Scholar] [CrossRef]
- Rothblat, D.S.; Schneider, J.S. Effects of GM1 ganglioside treatment on dopamine innervation of the striatum of MPTP-treated mice. Ann. N. Y. Acad. Sci. 1998, 845, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Kean, A.; DiStefano, L. GM1 ganglioside rescues substantia nigra pars compacta neurons and increases dopamine synthesis in residual nigrostriatal dopaminergic neurons in MPTP-treated mice. J. Neurosci. Res. 1995, 42, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Kastner, A.; Herrero, M.T.; Hirsch, E.C.; Guillen, J.; Luquin, M.R.; Javoy-Agid, F.; Obeso, J.A.; Agid, Y. Decreased tyrosine hydroxylase content in the dopaminergic neurons of MPTP-intoxicated monkeys: Effect of levodopa and GM1 ganglioside therapy. Ann. Neurol. 1994, 36, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.T.; Kastner, A.; Perez-Otaño, I.; Hirsch, E.C.; Luquin, M.R.; Javoy-Agid, F.; Del Río, J.; Obeso, J.A.; Agid, Y. Gangliosides and parkinsonism. Neurology 1993, 43, 2132–2134. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Pope, A.; Simpson, K.; Taggart, J.; Smith, M.G.; DiStefano, L. Recovery from experimental parkinsonism in primates with GM1 ganglioside treatment. Science 1992, 256, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S. MPTP-induced parkinsonism: Acceleration of biochemical and behavioral recovery by GM1 ganglioside treatment. J. Neurosci. Res. 1992, 31, 112–119. [Google Scholar] [CrossRef]
- Fazzini, E.; Durso, R.; Davoudi, H.; Szabo, G.K.; Albert, M.L. GM1 gangliosides alter acute MPTP-induced behavioral and neurochemical toxicity in mice. J. Neurol. Sci. 1990, 99, 59–68. [Google Scholar] [CrossRef]
- Gupta, M.; Schwarz, J.; Chen, X.L.; Roisen, F.J. Gangliosides prevent MPTP toxicity in mice—An immunocytochemical study. Brain Res. 1990, 527, 330–334. [Google Scholar] [CrossRef]
- Schneider, J.S.; Yuwiler, A. GM1 ganglioside treatment promotes recovery of striatal dopamine concentrations in the mouse model of MPTP-induced parkinsonism. Exp. Neurol. 1989, 105, 177–183. [Google Scholar] [CrossRef]
- Hadjiconstantinou, M.; Mariani, A.P.; Neff, N.H. GM1 ganglioside-induced recovery of nigrostriatal dopaminergic neurons after MPTP: An immunohistochemical study. Brain Res. 1989, 484, 297–303. [Google Scholar] [CrossRef]
- Ba, X. Therapeutic effects of GM1 on Parkinson’s disease in rats and its mechanism. Int. J. Neurosci. 2016, 126, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Lundius, E.G.; Vukojevic, V.; Hertz, E.; Stroth, N.; Cederlund, A.; Hiraiwa, M.; Terenius, L.; Svennigsson, P. GPR37 protein trafficking to the plasma membrane regulated by prosaposin and GM1 gangliosides promotes cell viability. J. Biol. Chem. 2014, 289, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of alpha-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Garten, M.; Prévost, C.; Cadart, C.; Gautier, R.; Bousset, L.; Melki, R.; Bassereau, P.; Vanni, S. Methyl-branched lipids promote the membrane adsorption of α-synuclein by enhancing shallow lipid-packing defects. Phys. Chem. Chem. Phys. 2015, 17, 15589–15597. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef]
- Suzuki, K.; Iseki, E.; Togo, T.; Yamaguchi, A.; Katsuse, O.; Katsuyama, K.; Kanzaki, S.; Shiozaki, K.; Kawanishi, C.; Yamashita, S.; et al. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. 2007, 114, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Akkhawattanangkul, Y.; Maiti, P.; Xue, Y.; Aryal, D.; Wetsel, W.C.; Hamilton, D.; Fowler, S.C.; McDonald, M.P. Targeted deletion of GD3 synthase protects against MPTP-induced neurodegeneration. Genes Brain Behav. 2017, 16, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Shin, W.H.; Kim, J.; Joe, E.H.; Lee, Y.B.; Cho, K.G.; Oh, Y.J.; Kim, S.U.; Jun, B.K. Trisialoganglioside GT1b induces in vivo degeneration of nigral dopaminergic neurons: Role of microglia. Glia 2002, 38, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Sekigawa, A.; Sugama, S.; Takenouchi, T.; Masliah, E.; Hashimoto, M. Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Am. J. Pathol. 2009, 174, 1891–1909. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Finkielstein, C.V.; Capelluto, D.G.S. The enigmatic role of sulfatides: New insights into cellular functions and mechanisms of protein recognition. Adv. Exp. Med. Biol. 2013, 991, 27–40. [Google Scholar] [CrossRef]
- Antelmi, E.; Rizzo, G.; Fabbri, M.; Capellari, S.; Scaglione, C.; Martinelli, P. Arylsulphatase A activity in familial parkinsonism: A pathogenetic role? J. Neurol. 2014, 261, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Ippoliti, M.; Montanari, M.; Martinelli, A.; Mochi, M.; Giuliani, S.; Sangiorgi, S. Arylsulphatase A (ASA) activity in parkinsonism and symptomatic essential tremor. Acta Neurol. Scand. 1994, 89, 171–174. [Google Scholar] [CrossRef]
- Cheng, H.; Xu, J.; McKeel, D.W.; Han, X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer’s disease: An electrospray ionization mass spectrometric study. Cell. Mol. Biol. (Noisy-le-grand) 2003, 49, 809–818. [Google Scholar]
- Spann, N.J.; Glass, C.K. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013, 14, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hannich, J.T.; Umebayashi, K.; Riezman, H. Distribution and Functions of Sterols and Sphingolipids. Cold Spring Harb. Perspect. Biol. 2011, 3, a004762. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.M.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D.; Checkoway, H. Dietary fats, cholesterol and iron as risk factors for Parkinson’s disease. Park. Relat. Disord. 2009, 15, 47–52. [Google Scholar] [CrossRef]
- Johnson, C.C.; Gorell, J.M.; Rybicki, B.A.; Sanders, K.; Peterson, E.L. Adult nutrient intake as a risk factor for Parkinson’s disease. Int. J. Epidemiol. 1999, 28, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Sanderson, W.T.; Burchfiel, C.M.; Kashon, M.; Sharp, D.S.; Masaki, K.H.; Curb, J.D.; Petrovitch, H. Environmental, life-style, and physical precursors of clinical Parkinson’s disease: Recent findings from the Honolulu-Asia Aging Study. J. Neurol. 2003, 250 (Suppl. 3), III30–III39. [Google Scholar] [CrossRef]
- Wang, A.; Lin, Y.; Wu, Y.; Zhang, D. Macronutrients intake and risk of Parkinson’s disease: A meta-analysis. Geriatr. Gerontol. Int. 2015, 15, 606–616. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wang, M.; Sterling, N.W.; Du, G.; Lewis, M.M.; Yao, T.; Mailman, R.B.; Li, R.; Huang, X. Circulating Cholesterol Levels May Link to the Factors Influencing Parkinson’s Risk. Front. Neurol. 2017, 8, 501. [Google Scholar] [CrossRef]
- Kirbas, A.; Kirbas, S.; Cure, M.C.; Tufekci, A. Paraoxonase and arylesterase activity and total oxidative/anti-oxidative status in patients with idiopathic Parkinson’s disease. J. Clin. Neurosci. 2014, 21, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nakamura, Y.; Kiyozuka, T.; Aoyagi, J.; Hirayama, T.; Nagata, R.; Ito, H.; Iwamoto, K.; Murata, K.; Yoshii, Y.; et al. Serological profiles of urate, paraoxonase-1, ferritin and lipid in Parkinson’s disease: Changes linked to disease progression. Neurodegener. Dis. 2011, 8, 252–258. [Google Scholar] [CrossRef]
- Huang, X.; Alonso, A.; Guo, X.; Umbach, D.M.; Lichtenstein, M.L.; Ballantyne, C.M.; Mailman, R.B.; Mosley, T.H.; Chen, H. Statins, plasma cholesterol, and risk of Parkinson’s disease: A prospective study. Mov. Disord. 2015, 30, 552–559. [Google Scholar] [CrossRef]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Case-control study of risk of Parkinson’s disease in relation to hypertension, hypercholesterolemia, and diabetes in Japan. J. Neurol. Sci. 2010, 293, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Chen, H.; Schwarzschild, M.; Ascherio, A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 2007, 69, 1688–1695. [Google Scholar] [CrossRef]
- De Lau, L.M.L.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Serum cholesterol levels and the risk of Parkinson’s disease. Am. J. Epidemiol. 2006, 164, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Rozani, V.; Gurevich, T.; Giladi, N.; El-Ad, B.; Tsamir, J.; Hemo, B.; Peretz, C. Higher serum cholesterol and decreased Parkinson’s disease risk: A statin-free cohort study. Mov. Disord. 2018, 33, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Auinger, P.; Eberly, S.; Oakes, D.; Schwarzschild, M.; Ascherio, A.; Mailman, R.; Chen, H.; Parkinson Study Group DATATOP Investigators. Serum cholesterol the progression of Parkinson’s disease: Results from DATATOP. PLoS ONE 2011, 6, e22854. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Bansal, D.; Muthyala, H. Role of serum cholesterol in Parkinson’s disease: A meta-analysis of evidence. J. Park. Dis. 2013, 3, 363–370. [Google Scholar] [CrossRef]
- Sterling, N.W.; Lichtenstein, M.; Lee, E.Y.; Lewis, M.M.; Evans, A.; Eslinger, P.J.; Du, G.; Gao, X.; Chen, H.; Kong, L.; et al. Higher Plasma LDL-Cholesterol is Associated with Preserved Executive and Fine Motor Functions in Parkinson’s Disease. Aging Dis. 2016, 7, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Ahlskog, J.E.; Rocca, W.A. Metabolic markers or conditions preceding Parkinson’s disease: A case-control study. Mov. Disord. 2012, 27, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Banerjee, B.D.; Bala, K.; Mitrabasu Dung Dung, A.A.; Chhillar, N. APOE and LRPAP1 gene polymorphism and risk of Parkinson’s disease. Neurol. Sci. 2014, 35, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Trautmann, E.; Sixel-Döring, F.; Wicke, T.; Ebentheuer, J.; Schaumburg, M.; Lang, E.; Focke, N.K.; Kumar, K.R.; Lohmann, K.; et al. Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology 2013, 81, 1226–1234. [Google Scholar] [CrossRef]
- Hu, G.; Antikainen, R.; Jousilahti, P.; Kivipelto, M.; Tuomilehto, J. Total cholesterol and the risk of Parkinson disease. Neurology 2008, 70, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Cassani, E.; Barichella, M.; Caccialanza, R.; Pezzoli, G. Anthropometric indices of fat distribution and cardiometabolic risk in Parkinson’s disease. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Musanti, R.; Parati, E.; Lamperti, E.; Ghiselli, G. Decreased cholesterol biosynthesis in fibroblasts from patients with Parkinson disease. Biochem. Med. Metab. Biol. 1993, 49, 133–142. [Google Scholar] [CrossRef]
- Shulman, J.M.; Yu, L.; Buchman, A.S.; Evans, D.A.; Schneider, J.A.; Bennett, D.A.; De Jager, P.L. Association of Parkinson Disease Risk Loci With Mild Parkinsonian Signs in Older Persons. JAMA Neurol. 2014, 71, 429. [Google Scholar] [CrossRef] [PubMed]
- Lou, F.; Li, M.; Liu, N.; Li, X.; Ren, Y.; Luo, X. The Polymorphism of SREBF1 Gene rs11868035 G/A Is Associated with susceptibility to Parkinson’s disease in a Chinese Population. Int. J. Neurosci. 2018, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Cao, B.; Wu, Y.; Chen, Y.; Wei, Q.; Ou, R.; Yang, J.; Chen, X.; Zhao, B.; Song, W.; et al. Association analysis of SNP rs11868035 in SREBF1 with sporadic Parkinson’s disease, sporadic amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurosci. Lett. 2018, 664, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.A.; Fogel, A.I.; Wang, C.; MacArthur, R.; Guha, R.; Heman-Ackah, S.; Martin, S.; Youle, R.J.; Inglese, J. Chemogenomic profiling of endogenous PARK2 expression using a genome-edited coincidence reporter. ACS Chem. Biol. 2015, 10, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Yamane, T.; Takahashi-Niki, K.; Kato, I.; Niki, T.; Goldberg, M.S.; Shen, J.; Ishimoto, K.; Doi, T.; Iguchi-Ariga, S.M. Transcriptional activation of low-density lipoprotein receptor gene by DJ-1 and effect of DJ-1 on cholesterol homeostasis. PLoS ONE 2012, 7, e38144. [Google Scholar] [CrossRef]
- Kim, J.M.; Cha, S.H.; Choi, Y.R.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef]
- Kyung, J.W.; Kim, J.M.; Lee, W.; Ha, T.Y.; Cha, S.H.; Chung, K.H.; Choi, D.J.; Joi, I.; Song, W.K.; Joe, E.H.; et al. DJ-1 deficiency impairs synaptic vesicle endocytosis and reavailability at nerve terminals. Proc. Natl. Acad. Sci. USA 2018, 115, 1629–1634. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef]
- Cha, S.H.; Choi, Y.R.; Heo, C.H.; Kang, S.J.; Joe, E.H.; Jou, I.; Kim, H.M.; Park, S.M. Loss of parkin promotes lipid rafts-dependent endocytosis through accumulating caveolin-1: Implications for Parkinson’s disease. Mol. Neurodegener. 2015, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, P.; Orgaz, L.; Bueno-Gil, G.; Espadas, I.; Rodríguez-Traver, E.; Kulisevsky, J.; Gutierrez, A.; Dávila, J.C.; González-Polo, R.A.; Fuentes, J.M.; et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 2017, 32, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.A.S.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE 2013, 8, e80705. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, I.; Nath, S.; Bornefall, P.; Giraldo, A.M.V.; Öllinger, K. Impact of high cholesterol in a Parkinson’s disease model: Prevention of lysosomal leakage versus stimulation of α-synuclein aggregation. Eur. J. Cell Biol. 2017, 96, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dehay, B.; Bezard, E.; Garcia-Ladona, F.J. U18666A, an activator of sterol regulatory element binding protein pathway, modulates presynaptic dopaminergic phenotype of SH-SY5Y neuroblastoma cells. Synapse 2017, 71, e21980. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.; Morin, N.; Rouillard, C.; Di Paolo, T. Membrane cholesterol removal and replenishment affect rat and monkey brain monoamine transporters. Neuropharmacology 2018, 133, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Choudhury, A.; Kumar, S.; Giri, A.; Sandhir, R.; Borah, A. Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson’s disease: Involvement of mitochondrial dysfunctions and oxidative stress. PLoS ONE 2017, 12, e0171285. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Choudhury, A.; Chandra Boruah, D.; Devi, R.; Bhattacharya, P.; Choudhury, M.D.; Borach, A. Hypercholesterolemia causes psychomotor abnormalities in mice and alterations in cortico-striatal biogenic amine neurotransmitters: Relevance to Parkinson’s disease. Neurochem. Int. 2017, 108, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Dutta, A.; Phukan, B.C.; Mazumder, M.K.; Justin-Thenmozhi, A.; Manivasagam, T.; Bhattacharya, P.; Borah, A. Accumulation of Cholesterol and Homocysteine in the Nigrostriatal Pathway of Brain Contributes to the Dopaminergic Neurodegeneration in Mice. Neuroscience 2018, 388, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Raju, A.; Jaisankar, P.; Borah, A.; Mohanakumar, K.P. 1-Methyl-4-Phenylpyridinium-Induced Death of Differentiated SH-SY5Y Neurons Is Potentiated by Cholesterol. Ann. Neurosci. 2018, 24, 243–251. [Google Scholar] [CrossRef]
- Fantini, J.; Carlus, D.; Yahi, N. The fusogenic tilted peptide (67-78) of α-synuclein is a cholesterol binding domain. Biochim. Biophys. Acta 2011, 1808, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Beyer, K. Binding of alpha-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006, 281, 9251–9259. [Google Scholar] [CrossRef] [PubMed]
- Van Maarschalkerweerd, A.; Vetri, V.; Vestergaard, B. Cholesterol facilitates interactions between α-synuclein oligomers and charge-neutral membranes. FEBS Lett. 2015, 589, 2661–2667. [Google Scholar] [CrossRef] [PubMed]
- Shvadchak, V.V.; Falomir-Lockhart, L.J.; Yushchenko, D.A.; Jovin, T.M. Specificity and kinetics of alpha-synuclein binding to model membranes determined with fluorescent excited state intramolecular proton transfer (ESIPT) probe. J. Biol. Chem. 2011, 286, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Bar-On, P.; Crews, L.; Koob, A.O.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 2008, 105, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. α-Synuclein-induced synapse damage in cultured neurons is mediated by cholesterol-sensitive activation of cytoplasmic phospholipase A2. Biomolecules 2015, 5, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Rockenstein, E.; Adame, A.; Ho, G.; Hashimoto, M.; Masliah, E. Effects of the cholesterol-lowering compound methyl-beta-cyclodextrin in models of alpha-synucleinopathy. J. Neurochem. 2006, 98, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.H.T.; Halliday, G.M.; Kim, W.S. α-Synuclein Regulates Neuronal Cholesterol Efflux. Molecules 2017, 22, 1769. [Google Scholar] [CrossRef] [PubMed]
- Leftin, A.; Job, C.; Beyer, K.; Brown, M.F. Solid-state 13C NMR reveals annealing of raft-like membranes containing cholesterol by the intrinsically disordered protein α-Synuclein. J. Mol. Biol. 2013, 425, 2973–2987. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Chesselet, M.F.; Fleming, S.; Mortazavi, F.; Meurers, B. Strengths and limitations of genetic mouse models of Parkinson’s disease. Park. Relat. Disord. 2008, 14 (Suppl. 2), S84–S87. [Google Scholar] [CrossRef]
- Sheng, Z.; Jia, X.; Kang, M. Statin use and risk of Parkinson’s disease: A meta-analysis. Behav. Brain Res. 2016, 309, 29–34. [Google Scholar] [CrossRef]
- Friedman, B.; Lahad, A.; Dresner, Y.; Vinker, S. Long-term statin use and the risk of Parkinson’s disease. Am. J. Manag. Care 2013, 19, 626–632. [Google Scholar]
- Gao, X.; Simon, K.C.; Schwarzschild, M.A.; Ascherio, A. Prospective study of statin use and risk of Parkinson disease. Arch. Neurol. 2012, 69, 380–384. [Google Scholar] [CrossRef]
- Wolozin, B.; Wang, S.W.; Li, N.C.; Lee, A.; Lee, T.A.; Kazis, L.E. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007, 5, 20. [Google Scholar] [CrossRef]
- Huang, X.; Chen, H.; Miller, W.C.; Mailman, R.B.; Woodard, J.L.; Chen, P.C.; Xiang, D.; Murrow, R.M.; Wand, Y.Z.; Poole, C. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov. Disord. 2007, 22, 22. [Google Scholar] [CrossRef]
- Wahner, A.D.; Bronstein, J.M.; Bordelon, Y.M.; Ritz, B. Statin use and the risk of Parkinson disease. Neurology 2008, 70, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Rozani, V.; Giladi, N.; El-Ad, B.; Gurevich, T.; Tsamir, J.; Hemo, B.; Peretz, C. Statin adherence and the risk of Parkinson’s disease: A population-based cohort study. PLoS ONE 2017, 12, e0175054. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Manthripragada, A.D.; Qian, L.; Schernhammer, E.; Wermuth, L.; Olsen, J.; Friss, S. Statin use and Parkinson’s disease in Denmark. Mov. Disord. 2010, 25, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of statins and the risk of Parkinson’s disease: A retrospective case-control study in the UK. Drug Saf. 2008, 31, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sterling, N.W.; Kong, L.; Lewis, N.M.; Mailman, R.B.; Chen, H.; Leslie, D.; Huang, X. Statins may facilitate Parkinson’s disease: Insight gained from a large, national claims database. Mov Disord 2017, 32, 913–917. [Google Scholar] [CrossRef]
- Bykov, K.; Yoshida, K.; Weisskopf, M.G.; Gagne, J.J. Confounding of the association between statins and Parkinson disease: Systematic review and meta-analysis. Pharmacoepidemiol. Drug Saf. 2017, 26, 294–300. [Google Scholar] [CrossRef]
- Marques, N.F.; Castro, A.A.; Mancini, G.; Rocha, F.L.; Santos, A.R.S.; Prediger, R.D.; De Bem, A.F.; Tasca, C.I. Atorvastatin Prevents Early Oxidative Events and Modulates Inflammatory Mediators in the Striatum Following Intranasal 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) Administration in Rats. Neurotox. Res. 2018, 33, 549–559. [Google Scholar] [CrossRef]
- Yan, J.Q.; Ma, Y.J.; Sun, J.C.; Bai, S.F.; Huang, L.N. Neuroprotective effect of lovastatin by inhibiting NMDA receptor1 in 6-hydroxydopamine treated PC12 cells. Int. J. Clin. Exp. Med. 2014, 7, 3313–3319. [Google Scholar]
- Jiang, P.; Gan, M.; Lin, W.L.; Yen, S.H.C. Nutrient deprivation induces α-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front. Aging Neurosci. 2014, 6, 268. [Google Scholar] [CrossRef]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Long, L.; Yan, J.Q.; Wei, L.; Pan, M.Q.; Gao, H.M.; Zhou, P.; Liu, M.; Zhu, C.S.; Tang, B.S.; et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci. Ther. 2013, 19, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, N.; Gupta, A.; Kalonia, H.; Mishra, J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012, 1471, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. Simvastatin prevents 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain. Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, X.N.; Wang, L.; Yenari, M.A.; Ying, W.; Goh, B.C.; Lee, H.S.; Wilder-Smith, E.P.; Wong, P.T. Effects of high dose of simvastatin on levels of dopamine and its reuptake in prefrontal cortex and striatum among SD rats. Neurosci. Lett. 2006, 408, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Kreisler, A.; Gelé, P.; Wiart, J.F.; Lhermitte, M.; Destée, A.; Bordet, R. Lipid-lowering drugs in the MPTP mouse model of Parkinson’s disease: Fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Schirris, T.J.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.M.; Brandt, U.; Koopman, W.J.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-Induced Myopathy Is Associated with Mitochondrial Complex III Inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Tanji, K.; Miki, Y.; Yamada, M.; Kakita, A.; Takahasi, H.; Utsumi, J.; Sasaki, H.; Wakabayashi, K. Isopentenyl diphosphate isomerase, a cholesterol synthesizing enzyme, is localized in Lewy bodies. Neuropathology 2015, 35, 432–440. [Google Scholar] [CrossRef]
- Kabuto, H.; Yamanushi, T.T.; Janjua, N.; Takayama, F.; Mankura, M. Effects of squalene/squalane on dopamine levels, antioxidant enzyme activity, and fatty acid composition in the striatum of Parkinson’s disease mouse model. J. Oleo Sci. 2013, 62, 21–28. [Google Scholar] [CrossRef]
- Lim, L.; Jackson-Lewis, V.; Wong, L.C.; Shui, G.H.; Goh, A.X.H.; Kesavapany, S.; Jenner, A.M.; Fivaz, M.; Przedborski, S.; Wenk, M.R. Lanosterol induces mitochondrial uncoupling and protects dopaminergic neurons from cell death in a model for Parkinson’s disease. Cell Death Differ. 2012, 19, 416–427. [Google Scholar] [CrossRef]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson’s disease. PLoS ONE 2012, 7, e38113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Glukhova, S.A.; Caldwell, K.A.; Caldwell, G.A. NCEH-1 modulates cholesterol metabolism and protects against α-synuclein toxicity in a C. elegans model of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3823–3836. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Lövgren-Sandblom, A.; Leoni, V.; Meaney, S.; Brodin, L.; Salveson, L.; Winge, K.; Pålhagen, S.; Svenningsson, P. Oxysterols and Parkinson’s disease: Evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci. Lett. 2013, 555, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Holley, A.E.; Flitter, W.D.; Slater, T.F.; Wells, F.R.; Daniel, S.E.; Lee, A.J.; Jenner, P.; Marsden, C.D. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: An HPLC and ESR study. Mov. Disord. 1994, 9, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Patra, K.; Boxer, A.L.; Svenningsson, P. 24S-Hydroxycholesterol Correlates with Tau and Is Increased in Cerebrospinal Fluid in Parkinson’s Disease and Corticobasal Syndrome. Front. Neurol. 2018, 9, 756. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; Monaco, A.; Pedone, C.; Tessitore, A.; De Mase, A.; Tedeschi, G.; Netti, P.A.; Abrescia, P. The level of 24-hydroxycholesteryl esters decreases in plasma of patients with Parkinson’s disease. Neurosci. Lett. 2018, 672, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rantham Prabhakara, J.P.; Feist, G.; Thomasson, S.; Thompson, A.; Schommer, E.; Ghribi, O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1722–1729. [Google Scholar] [CrossRef]
- Marwarha, G.; Rhen, T.; Schommer, T.; Ghribi, O. The oxysterol 27-hydroxycholesterol regulates α-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors—Relevance to Parkinson’s disease. J. Neurochem. 2011, 119, 1119–1136. [Google Scholar] [CrossRef]
- Cheng, D.; Kim, W.S.; Garner, B. Regulation of alpha-synuclein expression by liver X receptor ligands in vitro. Neuroreport 2008, 19, 1685–1689. [Google Scholar] [CrossRef]
- Schommer, J.; Marwarha, G.; Schommer, T.; Flick, T.; Lund, J.; Ghribi, O. 27-Hydroxycholesterol increases α-synuclein protein levels through proteasomal inhibition in human dopaminergic neurons. BMC Neurosci. 2018, 19, 17. [Google Scholar] [CrossRef]
- Emanuelsson, I.; Norlin, M. Protective effects of 27- and 24-hydroxycholesterol against staurosporine-induced cell death in undifferentiated neuroblastoma SH-SY5Y cells. Neurosci. Lett. 2012, 525, 44–48. [Google Scholar] [CrossRef]
- Theofilopoulos, S.; Wang, Y.; Kitambi, S.S.; Sacchetti, P.; Sousa, K.M.; Bodin, K.; Kirk, J.; Saltó, C.; Gustafsson, M.; Toledo, E.M.; et al. Brain endogenous liver X receptor ligands selectively promote midbrain neurogenesis. Nat. Chem. Biol. 2013, 9, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, P.; Sousa, K.M.; Hall, A.C.; Liste, I.; Steffensen, K.R.; Theofilopoulos, S.; Parish, C.L.; Hazenber, C.; Richter, L.A.; Hovatta, O.; et al. Liver X receptors and oxysterols promote ventral midbrain neurogenesis in vivo and in human embryonic stem cells. Cell Stem Cell 2009, 5, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, I. Recent advances in physiological lipoprotein metabolism. Clin. Chem. Lab. Med. 2014, 52, 1695–1727. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Li, D.; Chen, Y.E.; Moon, J.J.; Schwendeman, A. High-Density Lipoproteins: Nature’s Multifunctional Nanoparticles. ACS Nano 2016, 10, 3015–3041. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.R.; Berlyand, Y.; Xie, S.X.; Alcalay, R.N.; Chahine, L.M.; Chen-Plotkin, A.S. Plasma apolipoprotein A1 associates with age at onset and motor severity in early Parkinson’s disease patients. Mov. Disord. 2015, 30, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.R.; Li, K.; Unger, T.L.; Gallagher, M.D.; Van Deerlin, V.M.; Agarwal, P.; Leverenz, J.; Roberts, J.; Samii, A.; Gross, R.G.; et al. Lower plasma apolipoprotein A1 levels are found in Parkinson’s disease and associate with apolipoprotein A1 genotype. Mov. Disord. 2015, 30, 805–812. [Google Scholar] [CrossRef]
- Lu, W.; Wan, X.; Liu, B.; Rong, X.; Zhu, L.; Li, P.; Li, J.; Wang, L.; Cui, L.; Wang, X. Specific changes of serum proteins in Parkinson’s disease patients. PLoS ONE 2014, 9, e95684. [Google Scholar] [CrossRef]
- Qiang, J.K.; Wong, Y.C.; Siderowf, A.; Hurtig, H.I.; Xie, S.X.; Lee, V.M.Y.; Trojanowski, J.Q.; Yearout, D.; Leverenz, J.; Montine, T.J.; et al. Plasma apolipoprotein A1 as a biomarker for Parkinson disease. Ann. Neurol. 2013, 74, 119–127. [Google Scholar] [CrossRef]
- Cassani, E.; Cereda, E.; Barichella, M.; Madio, C.; Cancello, R.; Caccialanza, R.; Zini, M.; Cilia, R.; Pezzoli, G. Cardiometabolic factors and disease duration in patients with Parkinson’s disease. Nutrition 2013, 29, 1331–1335. [Google Scholar] [CrossRef]
- Kawata, M.; Nemoto, Y.; Asahina, M.; Moroo, I.; Shinomiya, M.; Yamada, T. Risk factors for cerebral arteriosclerosis in Parkinson’s disease. Park. Relat. Disord. 1996, 2, 75–79. [Google Scholar] [CrossRef]
- Du, G.; Lewis, M.M.; Shaffer, M.L.; Chen, H.; Yang, Q.X.; Mailman, R.B.; Huang, X. Serum cholesterol and nigrostriatal R2* values in Parkinson’s disease. PLoS ONE 2012, 7, e35397. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Abbott, R.D.; Petrovitch, H.; Mailman, R.B.; Ross, G.W. Low LDL cholesterol and increased risk of Parkinson’s disease: Prospective results from Honolulu-Asia Aging Study. Mov. Disord. 2008, 23, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Nordestgaard, B.G.; Frikke-Schmidt, R.; Tybjærg-Hansen, A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: Mendelian randomisation study. BMJ 2017, 357, j1648. [Google Scholar] [CrossRef] [PubMed]
- Andican, G.; Konukoglu, D.; Bozluolcay, M.; Bayülkem, K.; Firtiına, S.; Burcak, G. Plasma oxidative and inflammatory markers in patients with idiopathic Parkinson’s disease. Acta Neurol. Belg. 2012, 112, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, H.; Williams, R.J.; Matin, R.; Iversen, L.; Rice-Evans, C.A. Phenolic antioxidants attenuate neuronal cell death following uptake of oxidized low-density lipoprotein. Free Radic. Biol. Med. 2000, 29, 1222–1233. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Tumanov, S.; Kamphorst, J.J. Recent advances in expanding the coverage of the lipidome. Curr. Opin. Biotechnol. 2017, 43, 127–133. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef]
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef]
- Andersen, O.S.; Koeppe, R.E. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Rohrbough, J.; Broadie, K. Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 2005, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Esposito, G.; Ana Clara, F.; Verstreken, P. Synaptic vesicle trafficking and Parkinson’s disease. Dev. Neurobiol. 2012, 72, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.M.; Heutink, P.; van de Berg, W.D. Evidence for Immune Response, Axonal Dysfunction and Reduced Endocytosis in the Substantia Nigra in Early Stage Parkinson’s Disease. PLoS ONE 2015, 10, e0128651. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef]
- Rustam, Y.H.; Reid, G.E. Analytical Challenges and Recent Advances in Mass Spectrometry Based Lipidomics. Anal. Chem. 2018, 90, 374–397. [Google Scholar] [CrossRef]
- Bou Khalil, M.; Hou, W.; Zhou, H.; Elisma, F.; Swayne, L.A.; Blanchard, A.P.; Yao, Z.; Bennett, S.A.; Figeys, D. Lipidomics era: Accomplishments and challenges. Mass Spectrom. Rev. 2010, 29, 877–929. [Google Scholar] [CrossRef]
- Hu, P.; Fabyanic, E.; Kwon, D.Y.; Tang, S.; Zhou, Z.; Wu, H. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol. Cell 2017, 68, 1006–1015.e7. [Google Scholar] [CrossRef] [PubMed]
- Rubakhin, S.S.; Romanova, E.V.; Nemes, P.; Sweedler, J.V. Profiling metabolites and peptides in single cells. Nat. Methods 2011, 8, S20–S29. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Bäckhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hoovield, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef]
- Caesar, R.; Nygren, H.; Orešič, M.; Bäckhed, F. Interaction between dietary lipids and gut microbiota regulates hepatic cholesterol metabolism. J. Lipid Res. 2016, 57, 474–481. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Minato, T.; Maeda, T.; Fujisawa, Y.; Tsuji, H.; Nomoto, K.; Ohno, K.; Hirayama, M. Progression of Parkinson’s disease is associated with gut dysbiosis: Two-year follow-up study. PLoS ONE 2017, 12, e0187307. [Google Scholar] [CrossRef] [PubMed]
- Cassani, E.; Barichella, M.; Cancello, R.; Cavanna, F.; Iorio, L.; Cereda, E.; Bolliri, C.; Zampella Maria, P.; Bianchi, F.; Cestaro, B.; et al. Increased urinary indoxyl sulfate (indican): New insights into gut dysbiosis in Parkinson’s disease. Park. Relat. Disord. 2015, 21, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lista, S.; Khachaturian, Z.S.; Rujescu, D.; Garaci, F.; Dubois, B.; Hampel, H. Application of Systems Theory in Longitudinal Studies on the Origin and Progression of Alzheimer’s Disease. Methods Mol. Biol. 2016, 1303, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Dehairs, J.; Derua, R.; Rueda-Rincon, N.; Swinnen, J.V. Lipidomics in drug development. Drug Discov. Today Technol. 2015, 13, 33–38. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. https://doi.org/10.3390/cells8010027
Xicoy H, Wieringa B, Martens GJM. The Role of Lipids in Parkinson’s Disease. Cells. 2019; 8(1):27. https://doi.org/10.3390/cells8010027
Chicago/Turabian StyleXicoy, Helena, Bé Wieringa, and Gerard J. M. Martens. 2019. "The Role of Lipids in Parkinson’s Disease" Cells 8, no. 1: 27. https://doi.org/10.3390/cells8010027
APA StyleXicoy, H., Wieringa, B., & Martens, G. J. M. (2019). The Role of Lipids in Parkinson’s Disease. Cells, 8(1), 27. https://doi.org/10.3390/cells8010027