Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. DNA Constructs
2.3. Lentivirus Preparation and Infection
2.4. Cell lysis and Western Blot Analysis
2.5. Immunofluorescence
2.6. Migration Assay
2.7. Matrigel Invasion Assay
2.8. Soft Agar Assay
2.9. Experimental In Vivo Metastasis Assay
2.10. Statistical Analysis
3. Results
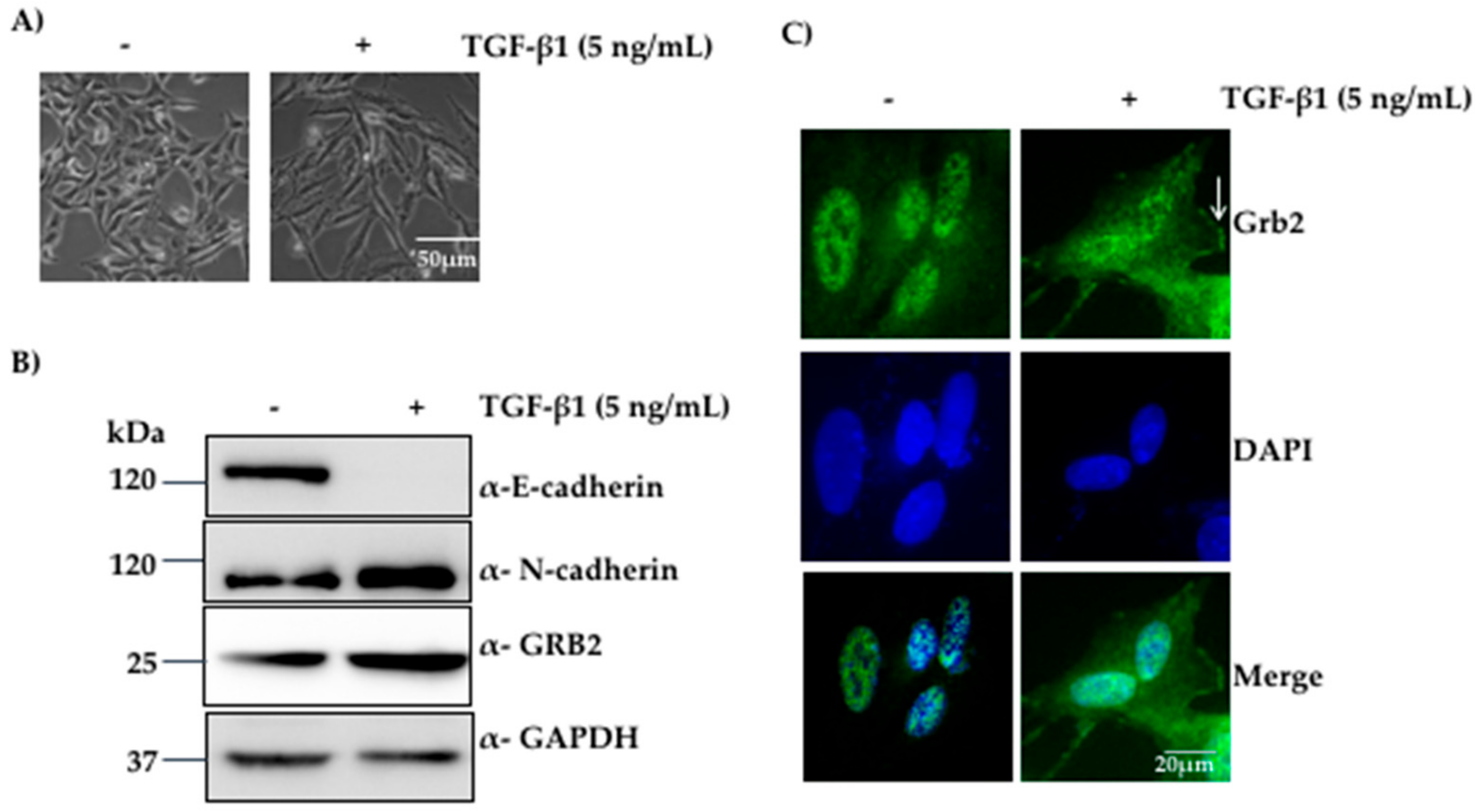
3.1. Expression of GRB2 Was Elevated during TGF-β1-Induced EMT in A549 Cells
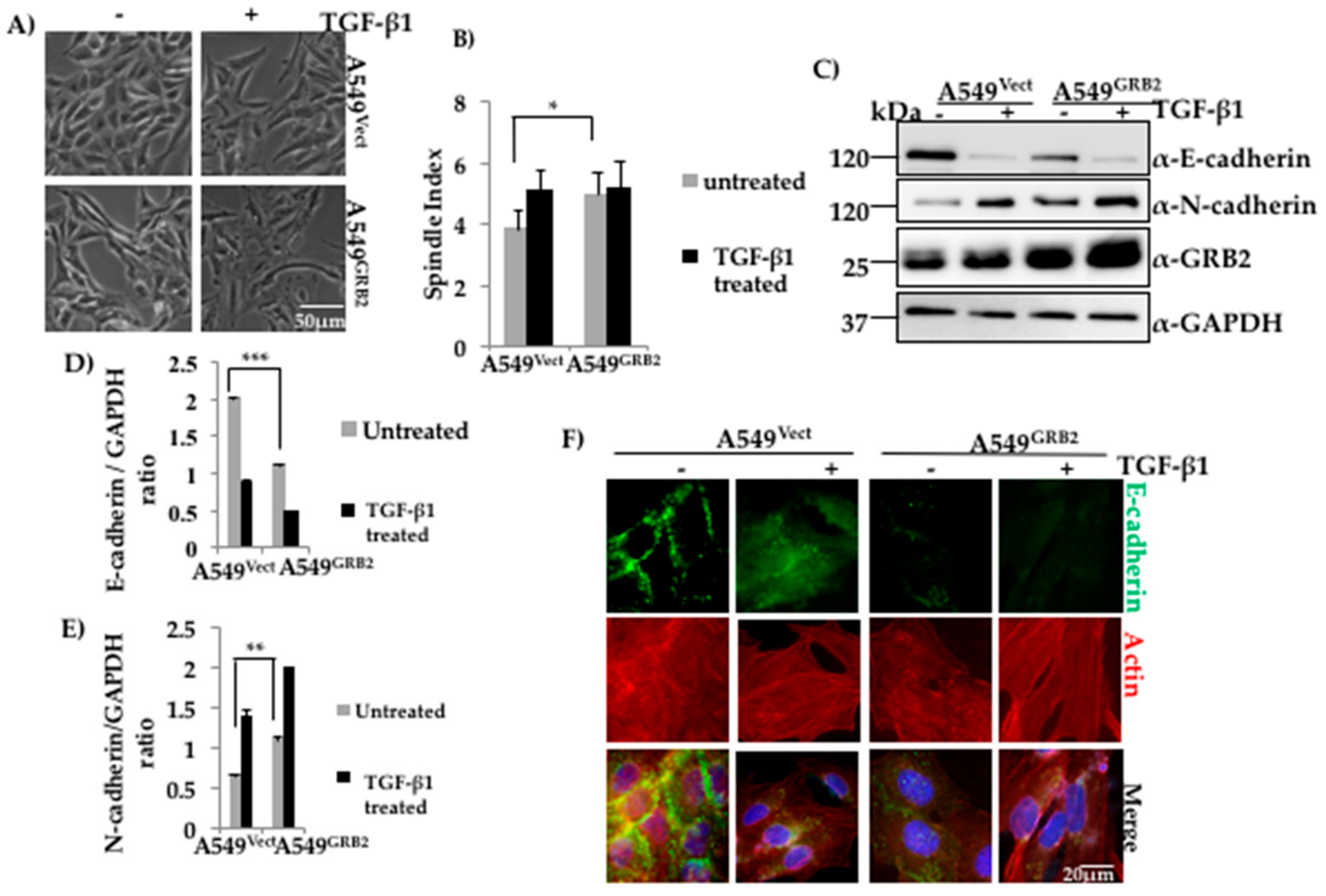
3.2. Overexpression of GRB2 Enhanced TGF-β1-Induced EMT in A549 Cells
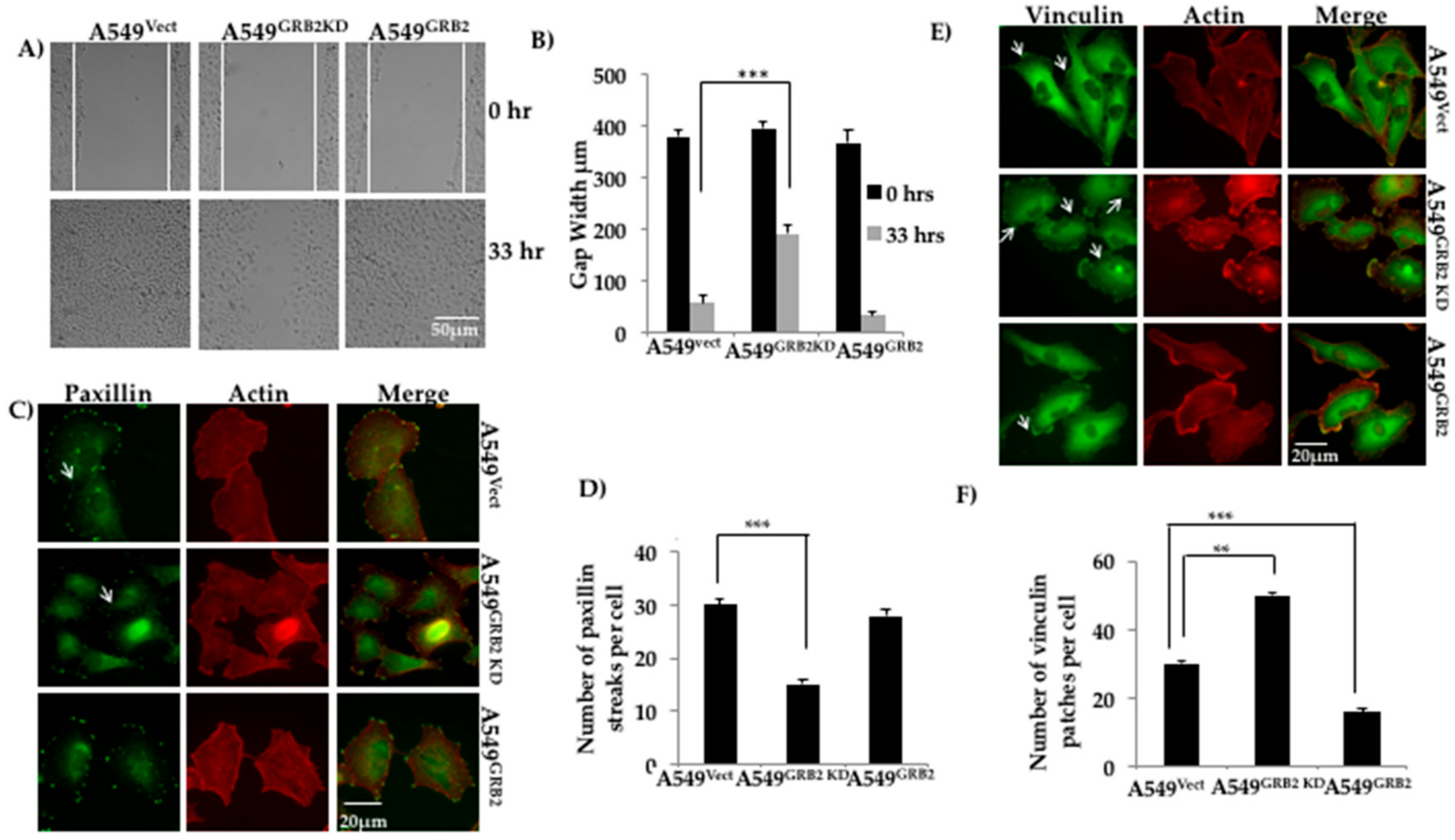
3.3. GRB2 Promotes Cell Migration and Reduced Vinculin Localisation in A549 Cells
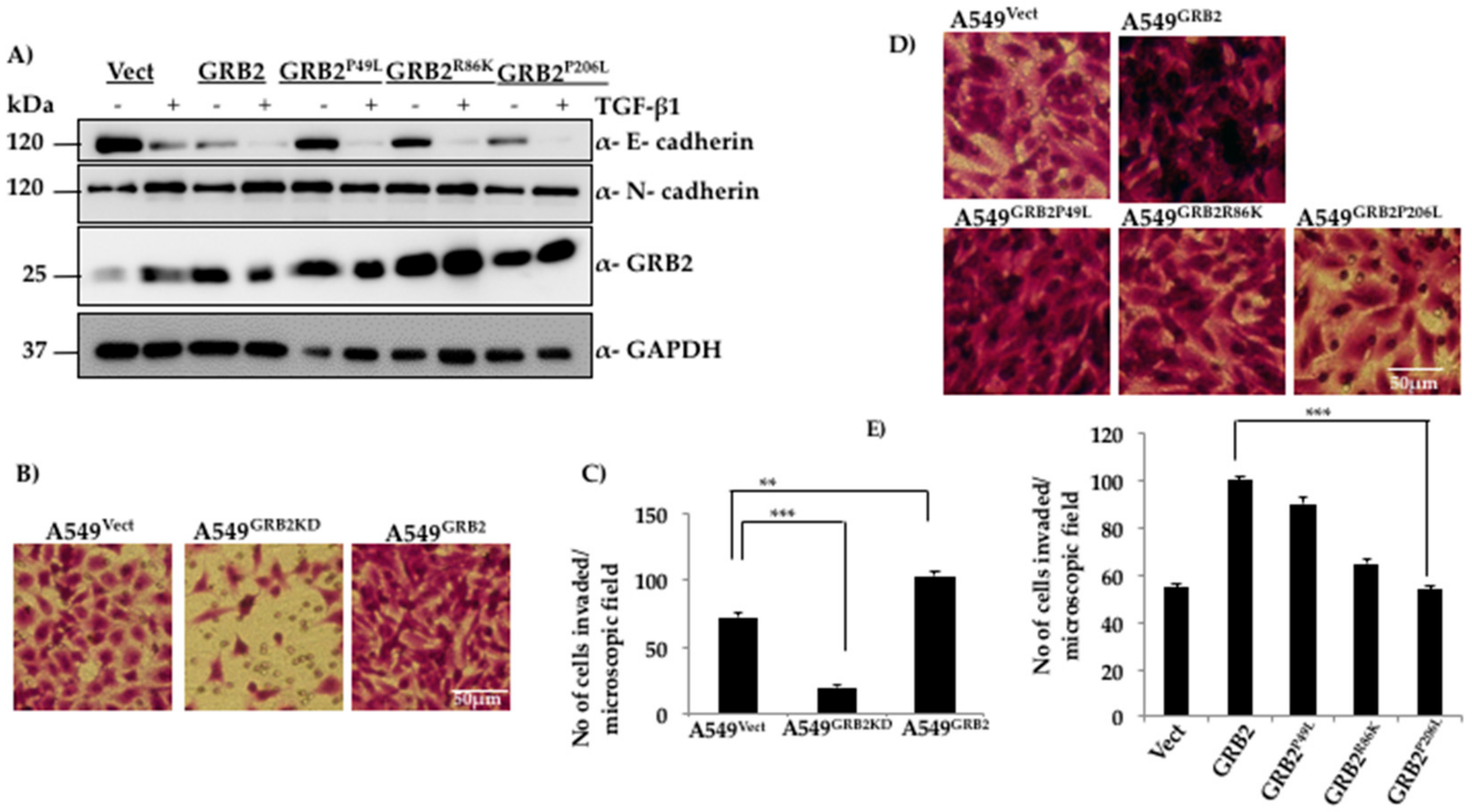
3.4. N-Terminal SH3 Domain and Central SH2 Domain of GRB2 Are Critical for Suppressing E-Cadherin Expression
3.5. C-Terminal SH3 Domain and Central SH2 Domain of GRB2 Is Critical for Promoting Invasion of A549 Cells
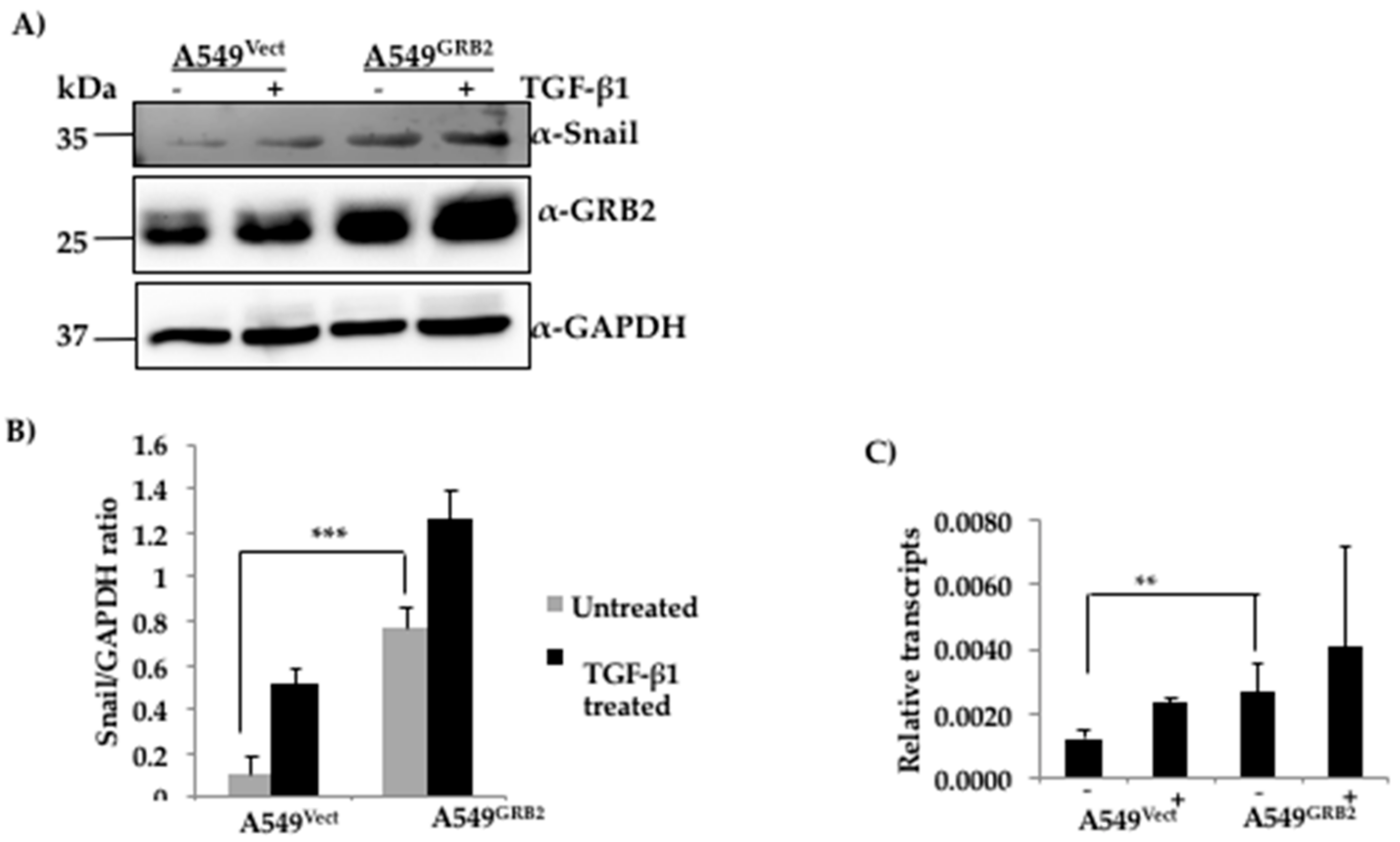
3.6. The Expression of SNAIL Is Increased in GRB2-Overexpressing A549 Cells
3.7. Overexpression of GRB2 Enhanced Anchorage-Independent Growth of A549 Cells
3.8. Overexpression of GRB2 Enhances In Vivo Lung Metastasis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, H.; Kang, Y. Multilayer control of the emt master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Giubellino, A.; Burke, T.R., Jr.; Bottaro, D.P. GRB2 signaling in cell motility and cancer. Expert Opin. Ther. Targets 2008, 12, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes. Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Ben-Ze’ev, A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol. Med. 2008, 14, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P. TGF-beta signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. SMADS orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. SMAD-dependent and SMAD-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-SMAD TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Emergence of the phosphoinositide 3-kinase-akt-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs 2011, 193, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R. Role of c-Met in cancer: Emphasis on lung cancer. Semin. Oncol. 2009, 36, S52–S58. [Google Scholar] [CrossRef] [PubMed]
- Chardin, P.; Cussac, D.; Maignan, S.; Ducruix, A. The GRB2 adaptor. FEBS Lett. 1995, 369, 47–51. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Rotin, D.; Batzer, A.; Mandiyan, V.; Schlessinger, J. SH3 domains direct cellular localization of signaling molecules. Cell 1993, 74, 83–91. [Google Scholar] [CrossRef]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.F.; Nioche, P.; Broutin-L’Hermite, I.; Boujemaa, R.; Le Clainche, C.; Egile, C.; Garbay, C.; Ducruix, A.; Sansonetti, P.; Pantaloni, D. GRB2 links signaling to actin assembly by enhancing interaction of Neural Wiskott-Aldrich syndrome protein (N-WASP) with actin-related protein (Arp2/3) complex. J. Biol. Chem. 2000, 275, 21946–21952. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 mapk during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef] [PubMed]
- Galliher-Beckley, A.J.; Schiemann, W.P. GRB2 binding to Tyr284 in TβR-II is essential for mammary tumor growth and metastasis stimulated by TGF-beta. Carcinogenesis 2008, 29, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates ERK map kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, B.S.; Adriaansen-Slot, S.S.; Rijksen, G.; Vroom, T.M. GRB2 overexpression in nuclei and cytoplasm of human breast cells: A histochemical and biochemical study of normal and neoplastic mammary tissue specimens. J. Pathol. 1997, 183, 195–203. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition—A hallmark of breast cancer metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Pandey, C.; Sze, S.K.; Thanabalu, T. Hypoxia activated EGFR signaling induces epithelial to mesenchymal transition (EMT). PLoS ONE 2012, 7, e49766. [Google Scholar] [CrossRef] [PubMed]
- Carisey, A.; Ballestrem, C. Vinculin, an adapter protein in control of cell adhesion signalling. Eur. J. Cell Biol. 2011, 90, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Lim, R.P.; Wu, Z.; Thanabalu, T. N-WASP plays a critical role in fibroblast adhesion and spreading. Biochem. Biophys. Res. Commun. 2007, 364, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, B.; Wyckoff, J.; Yamaguchi, H.; Wang, Y.; Roussos, E.T.; Condeelis, J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 2012, 125, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing e-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Dimitri, C.A.; Yoon, S.O.; Dowdle, W.; Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via def motif-dependent signaling events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Winitthana, T.; Lawanprasert, S.; Chanvorachote, P. Triclosan potentiates epithelial-to-mesenchymal transition in anoikis-resistant human lung cancer cells. PLoS ONE 2014, 9, e110851. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-beta signaling during tumor progression. Cancer Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Tanaka, M.; Uekita, T.; Inoue, S.; Yokota, J.; Ouchi, Y.; Sakai, R. Tyrosine phosphorylation of paxillin affects the metastatic potential of human osteosarcoma. Oncogene 2005, 24, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The role of snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, P.; Kalailingam, P.; Tan, H.B.; Thanabalu, T. Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression. Cells 2018, 7, 97. https://doi.org/10.3390/cells7080097
Mitra P, Kalailingam P, Tan HB, Thanabalu T. Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression. Cells. 2018; 7(8):97. https://doi.org/10.3390/cells7080097
Chicago/Turabian StyleMitra, Payal, Pazhanichamy Kalailingam, Hui Bing Tan, and Thirumaran Thanabalu. 2018. "Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression" Cells 7, no. 8: 97. https://doi.org/10.3390/cells7080097
APA StyleMitra, P., Kalailingam, P., Tan, H. B., & Thanabalu, T. (2018). Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression. Cells, 7(8), 97. https://doi.org/10.3390/cells7080097