Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
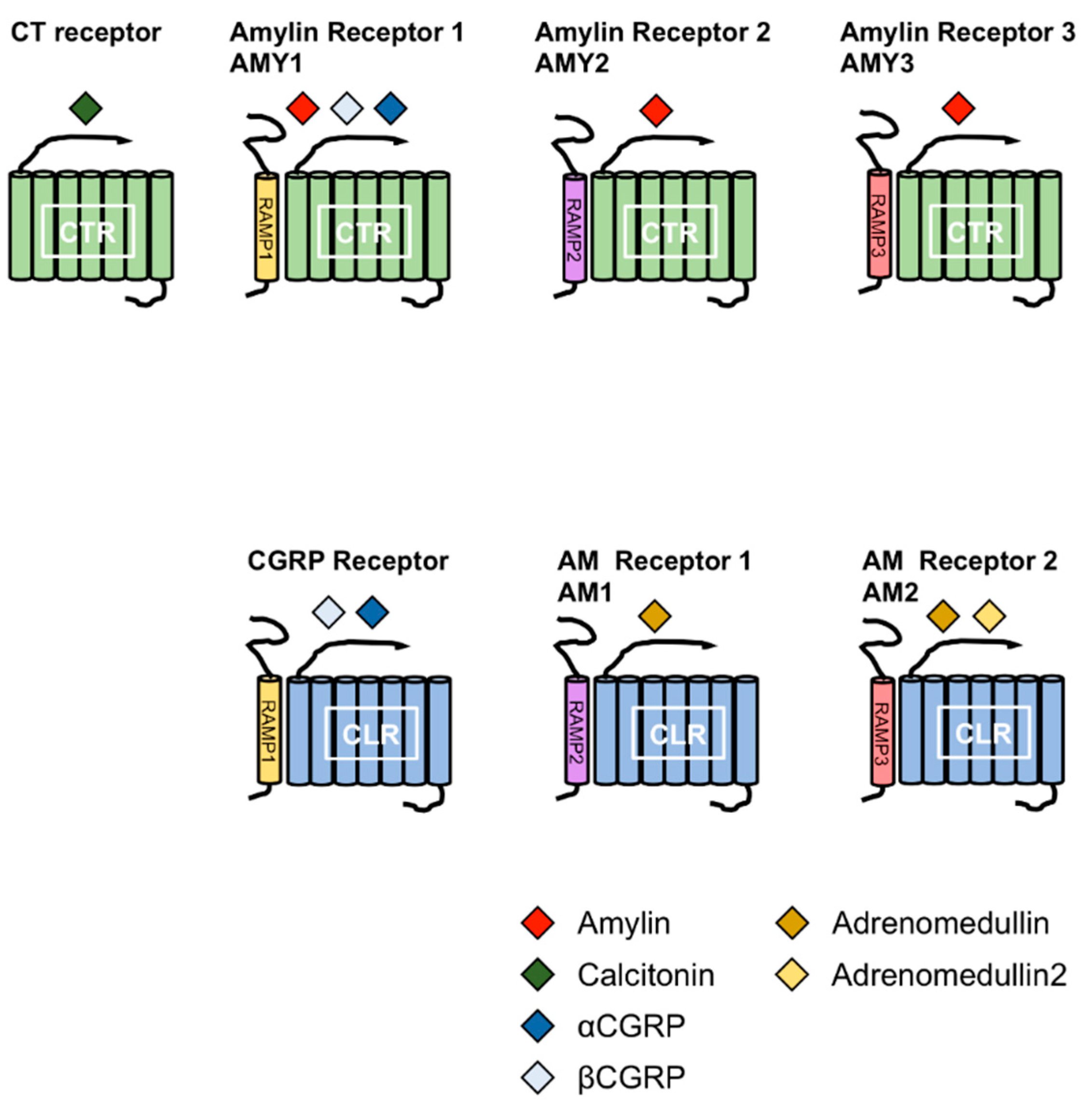
2. Physiological Role of Amylin
2.1. Role of Amylin in the Central Nervous System (CNS)
2.2. Role of Amylin in Pancreatic Islet β-Cells
3. Cytotoxicity of Amylin
4. Protection of Pancreatic Islet β-Cells from Cytotoxicity of Amylin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ogawa, A.; Harris, V.; McCorkle, S.K.; Unger, R.H.; Luskey, K.L. Amylin secretion from the rat pancreas and its selective loss after streptozotocin treatment. J. Clin. Investig. 1990, 85, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Cai, K.; Wang, O.; Li, Z.; Chen, J.; Deng, B.; Qian, L.; Le, Y. Fatty acids induce amylin expression and secretion by pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E99–E107. [Google Scholar] [CrossRef] [PubMed]
- Brereton, M.F.; Vergari, E.; Zhang, Q.; Clark, A. Alpha-, Delta- and PP-cells: Are They the Architectural Cornerstones of Islet Structure and Co-ordination? J. Histochem. Cytochem. 2015, 63, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Yada, T.; Damdindorj, B.; Rita, R.S.; Kurashina, T.; Ando, A.; Taguchi, M.; Koizumi, M.; Sone, H.; Nakata, M.; Kakei, M.; et al. Ghrelin signalling in beta-cells regulates insulin secretion and blood glucose. Diabetes Obes. Metab. 2014, 16 (Suppl. 1), 111–117. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J., Jr.; Porte, D., Jr. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 1990, 39, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Lukinius, A.; Wilander, E.; Westermark, G.T.; Engstrom, U.; Westermark, P. Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia 1989, 32, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Stridsberg, M.; Sandler, S.; Wilander, E. Cosecretion of islet amyloid polypeptide (IAPP) and insulin from isolated rat pancreatic islets following stimulation or inhibition of beta-cell function. Regul. Pept. 1993, 45, 363–370. [Google Scholar] [CrossRef]
- Kim, K.-H.; Seong, B.L. Peptide amidation: Production of peptide hormonesin vivo andin vitro. Biotechnol. Bioprocess Eng. 2001, 6, 244–251. [Google Scholar] [CrossRef]
- Marzban, L.; Soukhatcheva, G.; Verchere, C.B. Role of carboxypeptidase E in processing of pro-islet amyloid polypeptide in {beta}-cells. Endocrinology 2005, 146, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nomura, Y.; Tokumitsu, Y. Calcitonin gene expression induced by lipopolysaccharide in the rat pituitary. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1380–E1384. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Tsuchiya, H.; Murakami, T.; Satoh, K.; Tokumitsu, Y. Calcitonin induces IL-6 production via both PKA and PKC pathways in the pituitary folliculo-stellate cell line. Endocrinology 2001, 142, 3563–3569. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Garelja, M.L.; Poyner, D.R.; Walker, C.S. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 2018, 175, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sexton, P.M.; Houssami, S.; Hilton, J.M.; O’Keeffe, L.M.; Center, R.J.; Gillespie, M.T.; Darcy, P.; Findlay, D.M. Identification of brain isoforms of the rat calcitonin receptor. Mol. Endocrinol. 1993, 7, 815–821. [Google Scholar] [CrossRef] [PubMed]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Pioszak, A.A. Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: Molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 2004, 103, 21–80. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Borriello, M.; Carafa, V.; Altucci, L.; Vitiello, M.; Balestrieri, M.L.; Ricci, G.; Irace, G.; Sirangelo, I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim. Biophys. Acta 2016, 1862, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Giovane, A.; Maritato, R.; D’Onofrio, N.; Iannuzzi, C.; Giordano, A.; Irace, G.; Balestrieri, M.L. Platelet-activating factor mediates the cytotoxicity induced by W7FW14F apomyoglobin amyloid aggregates in neuroblastoma cells. J. Cell Biochem. 2014, 115, 2116–2122. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F. Zinc-ligand interactions modulate assembly and stability of the insulin hexamer—A review. Biometals 2005, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, Y.H.; O’Harte, F.P.; Barnett, C.R.; Flatt, P.R. Characterization of insulin glycation in insulin-secreting cells maintained in tissue culture. J. Endocrinol. 1997, 152, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Boland, C.; Meade, L.; Battise, D. Adjunctive therapy for glucose control in patients with type 1 diabetes. Diabetes Metab. Syndr. Obes. 2018, 11, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Del Prete, E.; Scharrer, E. Reduction of food intake in rats by intraperitoneal injection of low doses of amylin. Physiol. Behav. 1994, 55, 891–895. [Google Scholar] [CrossRef]
- Morley, J.E.; Flood, J.F.; Horowitz, M.; Morley, P.M.; Walter, M.J. Modulation of food intake by peripherally administered amylin. Am. J. Physiol. 1994, 267, R178–R184. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J. Differential permeability of the blood-brain barrier to two pancreatic peptides: Insulin and amylin. Peptides 1998, 19, 883–889. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Maness, L.M.; Huang, W.; Jaspan, J.B. Permeability of the blood-brain barrier to amylin. Life Sci. 1995, 57, 1993–2001. [Google Scholar] [CrossRef]
- Sexton, P.M.; Paxinos, G.; Kenney, M.A.; Wookey, P.J.; Beaumont, K. In vitro autoradiographic localization of amylin binding sites in rat brain. Neuroscience 1994, 62, 553–567. [Google Scholar] [CrossRef]
- Paxinos, G.; Chai, S.Y.; Christopoulos, G.; Huang, X.F.; Toga, A.W.; Wang, H.Q.; Sexton, P.M. In vitro autoradiographic localization of calcitonin and amylin binding sites in monkey brain. J. Chem. Neuroanat. 2004, 27, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Senn, M.; Althaus, J.; Del Prete, E.; Ehrensperger, F.; Scharrer, E. Lesion of the area postrema/nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides 1998, 19, 309–317. [Google Scholar] [CrossRef]
- Mollet, A.; Gilg, S.; Riediger, T.; Lutz, T.A. Infusion of the amylin antagonist AC 187 into the area postrema increases food intake in rats. Physiol. Behav. 2004, 81, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mietlicki-Baase, E.G.; Olivos, D.R.; Jeffrey, B.A.; Hayes, M.R. Cooperative interaction between leptin and amylin signaling in the ventral tegmental area for the control of food intake. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1116–E1122. [Google Scholar] [CrossRef] [PubMed]
- Mietlicki-Baase, E.G.; Rupprecht, L.E.; Olivos, D.R.; Zimmer, D.J.; Alter, M.D.; Pierce, R.C.; Schmidt, H.D.; Hayes, M.R. Amylin receptor signaling in the ventral tegmental area is physiologically relevant for the control of food intake. Neuropsychopharmacology 2013, 38, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Turek, V.F.; Trevaskis, J.L.; Levin, B.E.; Dunn-Meynell, A.A.; Irani, B.; Gu, G.; Wittmer, C.; Griffin, P.S.; Vu, C.; Parkes, D.G.; et al. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology 2010, 151, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Young, A.A.; Gedulin, B.; Vine, W.; Percy, A.; Rink, T.J. Gastric emptying is accelerated in diabetic BB rats and is slowed by subcutaneous injections of amylin. Diabetologia 1995, 38, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Clementi, G.; Caruso, A.; Cutuli, V.M.; de Bernardis, E.; Prato, A.; Amico-Roxas, M. Amylin given by central or peripheral routes decreases gastric emptying and intestinal transit in the rat. Experientia 1996, 52, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Reidelberger, R.D.; Arnelo, U.; Granqvist, L.; Permert, J. Comparative effects of amylin and cholecystokinin on food intake and gastric emptying in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R605–R611. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Lee, J.S.; Camilleri, M.; Szarka, L.A.; Burton, D.D.; Zinsmeister, A.R.; Rizza, R.A.; Klein, P.D. Effects of pramlintide, an amylin analogue, on gastric emptying in type 1 and 2 diabetes mellitus. Neurogastroenterol. Motil. 2002, 14, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.F.; King, P.; Macdonald, I.A.; Stubbs, T.A.; Perkins, A.C.; Blackshaw, P.E.; Moyses, C.; Tattersall, R.B. Infusion of pramlintide, a human amylin analogue, delays gastric emptying in men with IDDM. Diabetologia 1997, 40, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Samsom, M.; Szarka, L.A.; Camilleri, M.; Vella, A.; Zinsmeister, A.R.; Rizza, R.A. Pramlintide, an amylin analog, selectively delays gastric emptying: Potential role of vagal inhibition. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 278, G946–G951. [Google Scholar] [CrossRef] [PubMed]
- Woerle, H.J.; Albrecht, M.; Linke, R.; Zschau, S.; Neumann, C.; Nicolaus, M.; Gerich, J.E.; Goke, B.; Schirra, J. Impaired hyperglycemia-induced delay in gastric emptying in patients with type 1 diabetes deficient for islet amyloid polypeptide. Diabetes Care 2008, 31, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Young, A. Inhibition of gastric emptying. Adv. Pharmacol. 2005, 52, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Friis-Hansen, L.; Wierup, N.; Rehfeld, J.F.; Sundler, F. Reduced ghrelin, islet amyloid polypeptide, and peptide YY expression in the stomach of gastrin-cholecystokinin knockout mice. Endocrinology 2005, 146, 4464–4471. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.D.; Reddy, S.; Sun, X.; Yang, Y.; Krissansen, G.W. Distribution of insulin mRNA transcripts within the human body. Biochem. Biophys. Res. Commun. 2014, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, R.; Smith, D.M.; Bloom, S.R. Investigation and characterization of binding sites for islet amyloid polypeptide in rat membranes. Endocrinology 1992, 130, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, C.; Johnson, M.D.; Dunn-Meynell, A.A.; Boyle, C.N.; Lutz, T.A.; Levin, B.E. Amylin-induced central IL-6 production enhances ventromedial hypothalamic leptin signaling. Diabetes 2015, 64, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Peelman, F.; Zabeau, L.; Moharana, K.; Savvides, S.N.; Tavernier, J. 20 years of leptin: Insights into signaling assemblies of the leptin receptor. J. Endocrinol. 2014, 223, T9–23. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.B.; Myers, M.G., Jr. 20 years of leptin: Connecting leptin signaling to biological function. J. Endocrinol. 2014, 223, T25–T35. [Google Scholar] [CrossRef] [PubMed]
- Gebre-Medhin, S.; Mulder, H.; Pekny, M.; Westermark, G.; Tornell, J.; Westermark, P.; Sundler, F.; Ahren, B.; Betsholtz, C. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem. Biophys. Res. Commun. 1998, 250, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Visa, M.; Alcarraz-Vizan, G.; Montane, J.; Cadavez, L.; Castano, C.; Villanueva-Penacarrillo, M.L.; Servitja, J.M.; Novials, A. Islet amyloid polypeptide exerts a novel autocrine action in beta-cell signaling and proliferation. FASEB J. 2015, 29, 2970–2979. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C.; Svensson, V.; Rorsman, F.; Engstrom, U.; Westermark, G.T.; Wilander, E.; Johnson, K.; Westermark, P. Islet amyloid polypeptide (IAPP):cDNA cloning and identification of an amyloidogenic region associated with the species-specific occurrence of age-related diabetes mellitus. Exp. Cell Res. 1989, 183, 484–493. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C.; Christmansson, L.; Engstrom, U.; Rorsman, F.; Svensson, V.; Johnson, K.H.; Westermark, P. Sequence divergence in a specific region of islet amyloid polypeptide (IAPP) explains differences in islet amyloid formation between species. FEBS Lett. 1989, 251, 261–264. [Google Scholar] [CrossRef]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Singh, S.; de Pablo, J.J. Effect of proline mutations on the monomer conformations of amylin. Biophys. J. 2013, 105, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.J.; Jobe, L.J.; Martin, R. Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin. Ther. 2005, 27, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Luca, S.; Yau, W.M.; Leapman, R.; Tycko, R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry 2007, 46, 13505–13522. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Middleton, C.T.; Singh, S.; Reddy, A.S.; Woys, A.M.; Strasfeld, D.B.; Marek, P.; Raleigh, D.P.; de Pablo, J.J.; Zanni, M.T.; et al. 2DIR spectroscopy of human amylin fibrils reflects stable beta-sheet structure. J. Am. Chem. Soc. 2011, 133, 16062–16071. [Google Scholar] [CrossRef] [PubMed]
- Bedrood, S.; Li, Y.; Isas, J.M.; Hegde, B.G.; Baxa, U.; Haworth, I.S.; Langen, R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012, 287, 5235–5241. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin). Protein Sci. 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Plesner, A.; Cao, P.; Ridgway, Z.; Zhang, J.; Tu, L.H.; Middleton, C.T.; Chao, B.; Sartori, D.J.; Meng, F.; et al. Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Soeller, W.C.; Butler, P.C. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: Evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 2003, 52, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Snell, J.M.; Sheftic, S.R.; Patil, S.M.; Daniels, S.B.; Kolling, F.W.; Alexandrescu, A.T. pH dependence of amylin fibrillization. Biochemistry 2014, 53, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Raleigh, D.P. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry 2005, 44, 16284–16291. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Hartman, K.; Reid, K.R.; Kennedy, R.T.; Ramamoorthy, A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry 2008, 47, 12680–12688. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, M.S.; Lin, Y.; Kinoshita, M.; Kanemura, S.; Itoh, D.; Sugiki, T.; Okumura, M.; Ramamoorthy, A.; Lee, Y.H. Impact of membrane curvature on amyloid aggregation. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, S.A.; Langen, R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; St Clair, J.R.; London, E.; Raleigh, D.P. Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry 2017, 56, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.; Yigittop, H.; Elgersma, R.C.; Rijkers, D.T.; Liskamp, R.M.; de Kruijff, B.; Hoppener, J.W.; Antoinette Killian, J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006, 356, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.A.; Loria, J.P.; Miranker, A.D. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol. 2009, 393, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, S.A.; Langen, R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 2007, 1768, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Apostolidou, M.; Jayasinghe, S.A.; Langen, R. Structure of alpha-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008, 283, 17205–17210. [Google Scholar] [CrossRef] [PubMed]
- Nanga, R.P.; Brender, J.R.; Vivekanandan, S.; Ramamoorthy, A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 2011, 1808, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, J.; Weise, K.; Opitz, N.; Winter, R. The effect of Abeta on IAPP aggregation in the presence of an isolated beta-cell membrane. J. Mol. Biol. 2012, 421, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Brender, J.R.; Lee, D.K.; Ramamoorthy, A. Phosphatidylethanolamine enhances amyloid fiber-dependent membrane fragmentation. Biochemistry 2012, 51, 7676–7684. [Google Scholar] [CrossRef] [PubMed]
- Sakagashira, S.; Sanke, T.; Hanabusa, T.; Shimomura, H.; Ohagi, S.; Kumagaye, K.Y.; Nakajima, K.; Nanjo, K. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996, 45, 1279–1281. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.T.; Entrup, L.; Templin, A.T.; Hogan, M.F.; Mellati, M.; Zraika, S.; Hull, R.L.; Kahn, S.E. The S20G substitution in hIAPP is more amyloidogenic and cytotoxic than wild-type hIAPP in mouse islets. Diabetologia 2016, 59, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Sakagashira, S.; Hiddinga, H.J.; Tateishi, K.; Sanke, T.; Hanabusa, T.; Nanjo, K.; Eberhardt, N.L. S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am. J. Pathol. 2000, 157, 2101–2109. [Google Scholar] [CrossRef]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
- Gurlo, T.; Ryazantsev, S.; Huang, C.J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Mirzabekov, T.A.; Lin, M.C.; Kagan, B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational switching within dynamic oligomers underpins toxic gain-of-function by diabetes-associated amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Exley, C.; House, E.; Patel, T.; Wu, L.; Fraser, P.E. Human pro-islet amyloid polypeptide (ProIAPP(1-48)) forms amyloid fibrils and amyloid spherulites in vitro. J. Inorg. Biochem. 2010, 104, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Andersson, A.; Westermark, P.; Westermark, G.T. Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 2006, 49, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Marzban, L.; Rhodes, C.J.; Steiner, D.F.; Haataja, L.; Halban, P.A.; Verchere, C.B. Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 2006, 55, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Gurlo, T.; Kayed, R.; Butler, A.E.; Haataja, L.; Glabe, C.G.; Butler, P.C. Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced beta-cell apoptosis in h-IAPP transgenic mice. Diabetes 2007, 56, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.L.; Miranker, A.D. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J. Mol. Biol. 2004, 335, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Nedumpully-Govindan, P.; Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Li, Z.C.; Westermark, G.T.; Leckstrom, A.; Steiner, D.F. Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett. 1996, 379, 203–206. [Google Scholar] [CrossRef]
- Bram, Y.; Frydman-Marom, A.; Yanai, I.; Gilead, S.; Shaltiel-Karyo, R.; Amdursky, N.; Gazit, E. Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes-associated specific antibodies. Sci. Rep. 2014, 4, 4267. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H. Transcribing beta-cell mitochondria in health and disease. Mol. Metab. 2017, 6, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011, 18, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.G.; Aguilar, A.G.; Burillo, J.; Oca, R.G.; Manca, M.A.; Novials, A.; Alcarraz-Vizan, G.; Guillen, C.; Benito, M. Pancreatic beta cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Ashcroft, A.E.; Radford, S.E. Small molecule probes of protein aggregation. Curr. Opin. Chem. Biol. 2017, 39, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Meng, F.; Raleigh, D.P. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J. Am. Chem. Soc. 2007, 129, 11300–11301. [Google Scholar] [CrossRef] [PubMed]
- Gilead, S.; Gazit, E. Inhibition of amyloid fibril formation by peptide analogues modified with alpha-aminoisobutyric acid. Angew. Chem. Int. Ed. Engl. 2004, 43, 4041–4044. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Misra, A.; Vaishnavi, T.S.; Thota, C.; Gupta, M.; Ramakumar, S.; Chauhan, V.S. Conformationally restricted short peptides inhibit human islet amyloid polypeptide (hIAPP) fibrillization. Chem. Commun. 2013, 49, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Kapurniotu, A.; Schmauder, A.; Tenidis, K. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J. Mol. Biol. 2002, 315, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Kalita, S.; Kalita, S.; Sukumar, P.; Mandal, B. Disaggregation of Amylin Aggregate by Novel Conformationally Restricted Aminobenzoic Acid containing alpha/beta and alpha/gamma Hybrid Peptidomimetics. Sci. Rep. 2017, 7, 40095. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, D.F.; Raleigh, D.P. Effects of sequential proline substitutions on amyloid formation by human amylin 20–29. Biochemistry 1999, 38, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Acharya, R.; Mishra, A.; Ramakumar, S.; Ahmed, F.; Chauhan, V.S. Dehydrophenylalanine (DeltaPhe) as a beta breaker: Extended structure terminated by a DeltaPhe-induced turn in the pentapeptide Boc-Phe1-Ala2-Ile3-DeltaPhe4-Ala5-OMe. Chembiochem 2008, 9, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Tenidis, K.; Waldner, M.; Bernhagen, J.; Fischle, W.; Bergmann, M.; Weber, M.; Merkle, M.L.; Voelter, W.; Brunner, H.; Kapurniotu, A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000, 295, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Montane, J.; de Pablo, S.; Castano, C.; Rodriguez-Comas, J.; Cadavez, L.; Obach, M.; Visa, M.; Alcarraz-Vizan, G.; Sanchez-Martinez, M.; Nonell-Canals, A.; et al. Amyloid-induced beta-cell dysfunction and islet inflammation are ameliorated by 4-phenylbutyrate (PBA) treatment. FASEB J. 2017, 31, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Estel, K.; Seeliger, J.; Friedrich, R.P.; Dogan, S.; Wanker, E.E.; Winter, R.; Ebbinghaus, S. Modulation of human IAPP fibrillation: Cosolutes, crowders and chaperones. Phys. Chem. Chem. Phys. 2015, 17, 8338–8348. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Birol, M.; Schlamadinger, D.E.; Wojcik, S.P.; Rhoades, E.; Miranker, A.D. Foldamer-mediated manipulation of a pre-amyloid toxin. Nat. Commun. 2016, 7, 11412. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiriyama, Y.; Nochi, H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity. Cells 2018, 7, 95. https://doi.org/10.3390/cells7080095
Kiriyama Y, Nochi H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity. Cells. 2018; 7(8):95. https://doi.org/10.3390/cells7080095
Chicago/Turabian StyleKiriyama, Yoshimitsu, and Hiromi Nochi. 2018. "Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity" Cells 7, no. 8: 95. https://doi.org/10.3390/cells7080095
APA StyleKiriyama, Y., & Nochi, H. (2018). Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity. Cells, 7(8), 95. https://doi.org/10.3390/cells7080095