A Mitochondrial Encoded Messenger at the Nucleus



Abstract
1. Introduction
2. Mitochondria-Derived Peptides
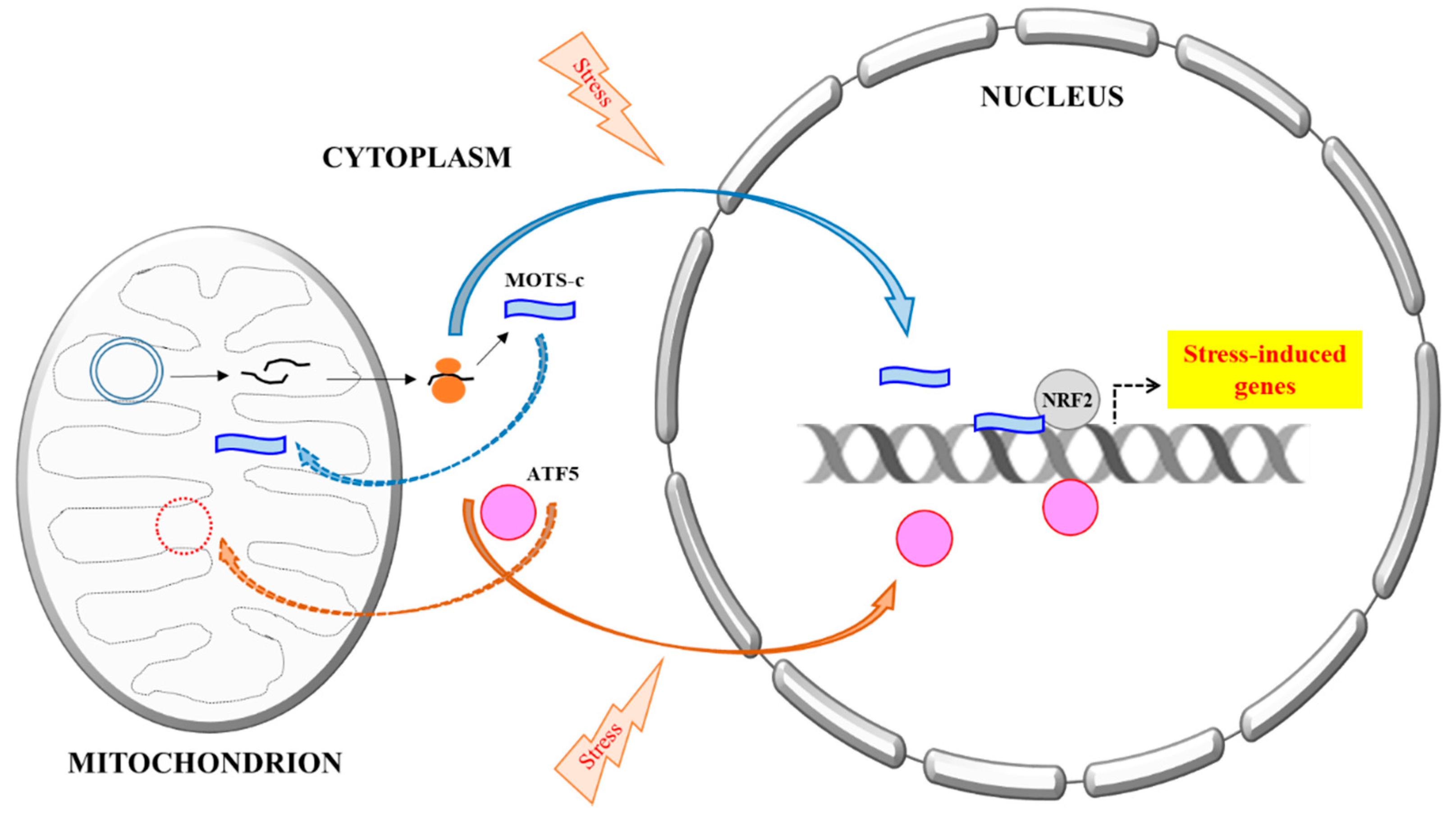
3. MOTS-c’s Mitochondrial–Nuclear Translocation and Activity
4. New Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Haynes, C.M. Integrating the UPRmt into the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Tan, T.M.C.; Takao, I.; Tang, B.L. The biochemistry and cell biology of aging: Metabolic regulation through mitochondrial signaling. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E581–E591. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, X.; Chen, P.; Liu, L.; Xin, N.; Tian, Y.; Dillin, A. The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 2018, 174, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Niikura, T.; Ito, Y.; Sudo, H.; Hata, M.; Arakawa, E.; Abe, Y.; Kita, Y.; Nishimoto, I. Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer’s disease-relevant insults. J. Neurosci. 2001, 21, 9235–9245. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Caricasole, A.; Bruno, V.; Cappuccio, I.; Melchiorri, D.; Copani, A.; Nicoletti, F. A novel rat gene encoding a Humanin-like peptide endowed with broad neuroprotective activity. FASEB J. 2002, 16, 1331–1333. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.T.; Huang, X.T.; Zhang, C.; Ke, Y. Humanin protects cortical neurons from ischemia and reperfusion injury by the increased activity of superoxide dismutase. Neurochem. Res. 2012, 37, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, R.H.; Huffman, D.M.; Calvert, J.W.; Jha, S.; Weinberg, Y.; Cui, L.; Nemkal, A.; Atzmon, G.; Klein, L.; Gundewar, S.; et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Thummasorn, S.; Apaijai, N.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc. Ther. 2016, 34, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The mitochondrial-derived peptide Humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Kuliawat, R.; Klein, L.; Gong, Z.; Nicoletta-Gentile, M.; Nemkal, A.; Cui, L.; Bastie, C.; Su, K.; Huffman, D.; Surana, M.; et al. Potent humanin analog increases glucose-stimulated insulin secretion through enhanced metabolism in the β cell. FASEB J. 2013, 27, 4890–4898. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Su, K.; Cui, L.; Tas, E.; Zhang, T.; Dong, H.H.; Yakar, S.; Muzumdar, R.H. Central effects of humanin on hepatic triglyceride secretion. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E283–E292. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Jin, J.; He, F.; Zheng, Y.; Li, T.; Zhang, Y.; He, J. Humanin promotes mitochondrial biogenesis in pancreatic MIN6 β-cells. Biochem. Biophys. Res. Commun. 2018, 497, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Maximov, V.; Martynenko, A.; Hunsmann, G.; Tarantul, V. Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer’s disease and target for cancer therapy. Med. Hypotheses 2002, 59, 670–673. [Google Scholar] [CrossRef]
- Bodzioch, M.; Lapicka-Bodzioch, K.; Zapala, B.; Kamysz, W.; Kiec-Wilk, B.; Dembinska-Kiec, A. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 2009, 94, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Iribarren, P.; Zhou, Y.; Gong, W.; Zhang, N.; Yu, Z.X.; Le, Y.; Cui, Y.; Wang, J.M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004, 172, 7078–7085. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Guerrero, N.; Wassef, G.; Xiao, J.; Mehta, H.H.; Cohen, P.; Yen, K. The mitochondrial-derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age-dependent signaling differences in the hippocampus. Oncotarget 2016, 7, 46899–46912. [Google Scholar] [CrossRef] [PubMed]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.K.; Teranishi, K.; Lobo, F.; Isas, J.M.; Xiao, J.; Yen, K.; Cohen, P.; Langen, R. The mitochondrial-derived peptides, HumaninS14G and small Humanin-like peptide 2, exhibit chaperone-like activity. Sci. Rep. 2017, 7, 7802. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Zarse, K.; Ristow, M. A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell Metab. 2015, 21, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gan, L.; Sha, X.; Lu, H.Y.; Jiang, Y.; Lei, X.Y.; Xu, C.; Ruan, B.J.; Wang, L.; Lu, Z.F. Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochem. Biophys. Res. Commun. 2016, 476, 412–419. [Google Scholar]
- Fuku, N.; Pareja-Galeano, H.; Zempo, H.; Alis, R.; Arai, Y.; Lucia, A.; Hirose, N. The mitochondrial-derived peptide MOTS-c: A player in exceptional longevity? Aging Cell 2015, 14, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhang, C.; Wu, W.; Liang, Y.; Wang, A.; Wu, S.; Zhao, Y.; Hou, L.; Ning, Q.; Luo, X. Circulating MOTS-c levels are decreased in obese male children and adolescents and associated with insulin resistance. Pediatr. Diabetes 2018. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Czypiorski, P.; Altschmied, J.; Rabanter, L.L.; Goy, C.; Jakob, S.; Haendeler, J. Outfielders playing in the infield: Functions of aging-associated “nuclear” proteins in the mitochondria. Curr. Mol. Med. 2014, 14, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, R.M.; Whitmarsh, A.J. Mitochondrial Proteins Moonlighting in the Nucleus. Trends Biochem. Sci. 2015, 40, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Lepore Signorile, M.; Longo, G.; De Rasmo, D.; et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.Y.; Leong, K.F.; Cronk, R.J.; Venkitachalam, S.; Pabich, S.; Yu, C.L. Nuclear localization of pyruvate dehydrogenase complex-E2 (PDC-E2), a mitochondrial enzyme, and its role in signal transducer and activator of transcription 5 (STAT5)-dependent gene transcription. Cell. Signal. 2011, 23, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, R.M.; Barnes, R.G.; Fisher, K.; Andreou, T.; Rooney, N.; Poulin, G.B.; Whitmarsh, A.J. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat. Cell Biol. 2015, 17, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Dacks, J.B.; Field, M.C.; Buick, R.; Eme, L.; Gribaldo, S.; Roger, A.J.; Brochier-Armanet, C.; Devos, D.P. The changing view of eukaryogenesis—Fossils, cells, lineages and how they all come together. J. Cell Sci. 2016, 129, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, F.; De Craemer, S.; Debunne, N.; Janssens, Y.; Wynendaele, E.; Van de Wiele, C.; De Spiegeleer, B. Peptides as quorum sensing molecules: Measurement techniques and obtained levels in vitro and in vivo. Front. Neurosci. 2017, 11, 183. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, Y.J.; Cai, Y.; Xu, M.H. The role of mitochondria in mTOR-regulated longevity. Biol. Rev. Camb. Philos. Soc. 2015, 90, 167–181. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name | Origin and Properties | Activities |
|---|---|---|
| Humanin | 21-aa (mitochondrial) or 24-aa (cytosol) peptide from open reading frame of mitochondrial 16S rRNA locus [16,17,18,28]. Also multiple humanin-like sequences from nuclear loci [29]. | |
| Rattin | 38-aa rat orthologue of human humanin [19] |
|
| Mitochondrial open reading frame of the 12S rRNA-c (MOTS-c) | 16-aa peptide from open reading frame of mitochondrial 12S rRNA locus [34] | |
| Small humanin-like peptide 1–6 (SHLP1–6) | 20–38-aa peptide from open reading frame of mitochondrial 16S rRNA locus [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yong, C.Q.Y.; Tang, B.L. A Mitochondrial Encoded Messenger at the Nucleus. Cells 2018, 7, 105. https://doi.org/10.3390/cells7080105
Yong CQY, Tang BL. A Mitochondrial Encoded Messenger at the Nucleus. Cells. 2018; 7(8):105. https://doi.org/10.3390/cells7080105
Chicago/Turabian StyleYong, Cheryl Qian Ying, and Bor Luen Tang. 2018. "A Mitochondrial Encoded Messenger at the Nucleus" Cells 7, no. 8: 105. https://doi.org/10.3390/cells7080105
APA StyleYong, C. Q. Y., & Tang, B. L. (2018). A Mitochondrial Encoded Messenger at the Nucleus. Cells, 7(8), 105. https://doi.org/10.3390/cells7080105