Tracking Cell Recruitment and Behavior within the Tumor Microenvironment Using Advanced Intravital Imaging Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Antibodies and Dyes
2.3. Cell Culture
2.4. Preparing Cells for Tumour Implantation
2.5. Tumor Implantation
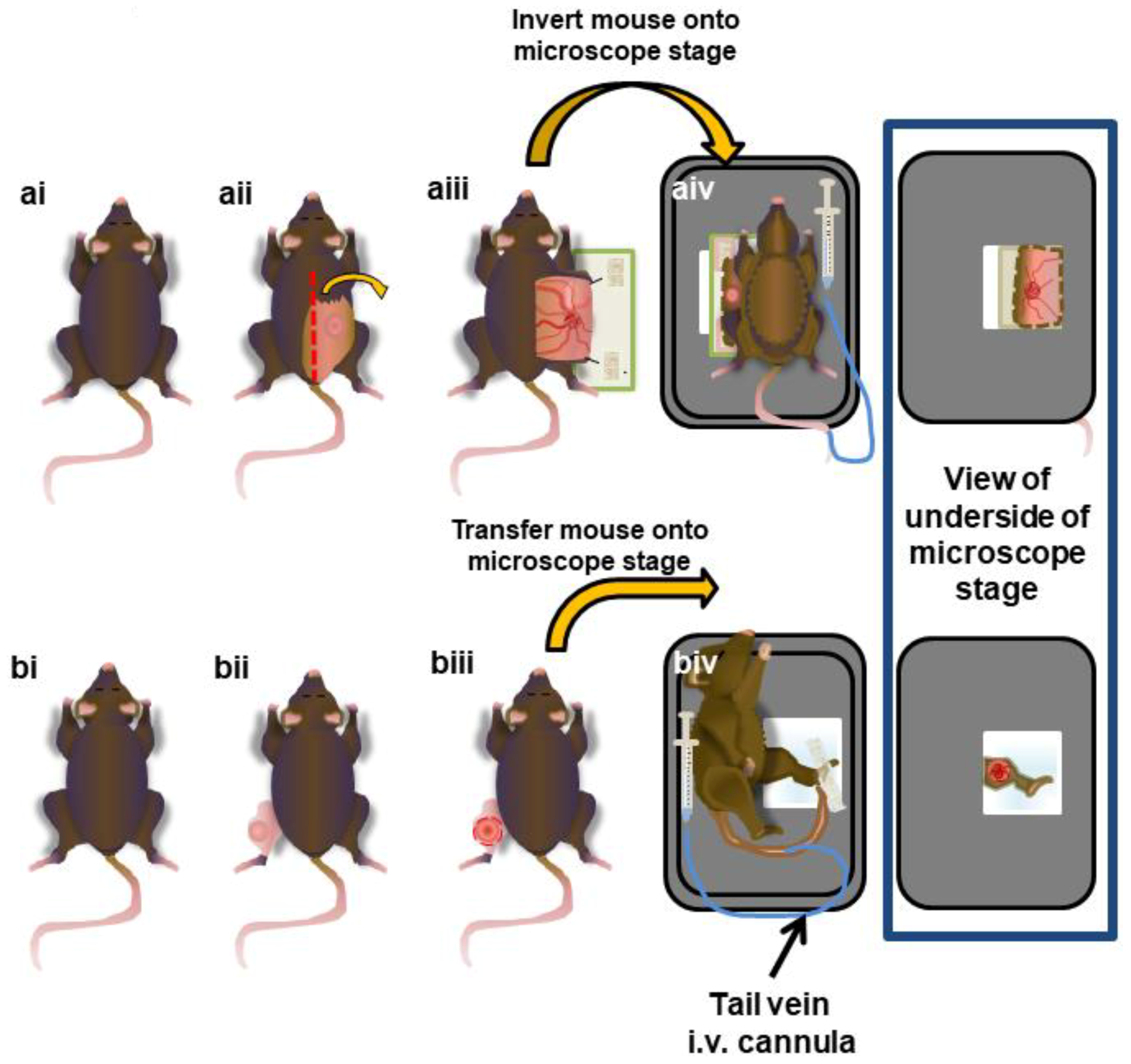
2.6. Surgical Preparation of Subcutaneous Tumours
2.7. Surgical Preparation of Intramuscular Tumors
2.8. IVM Imaging
2.9. Cell Tracking Analysis
3. Results
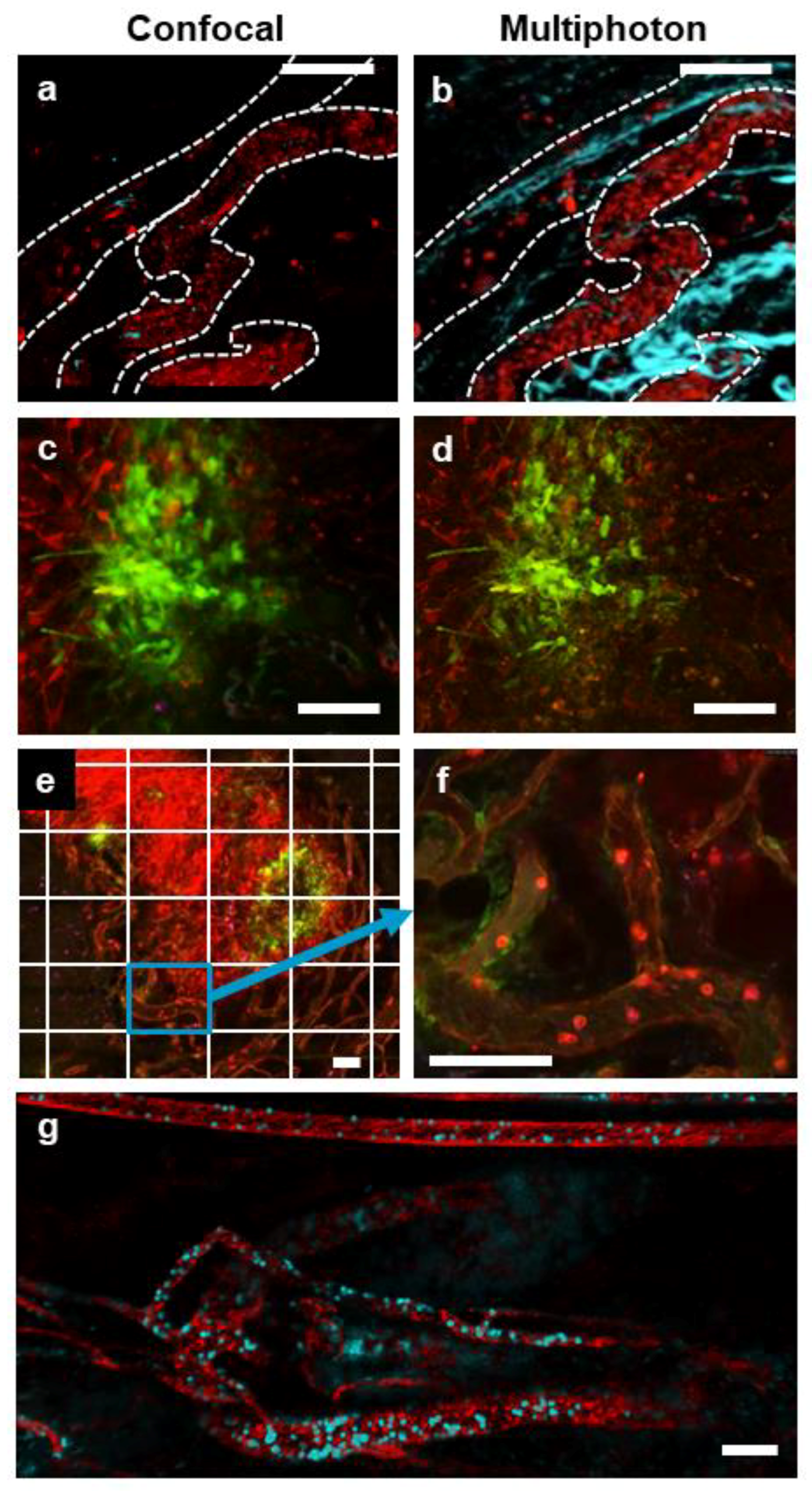
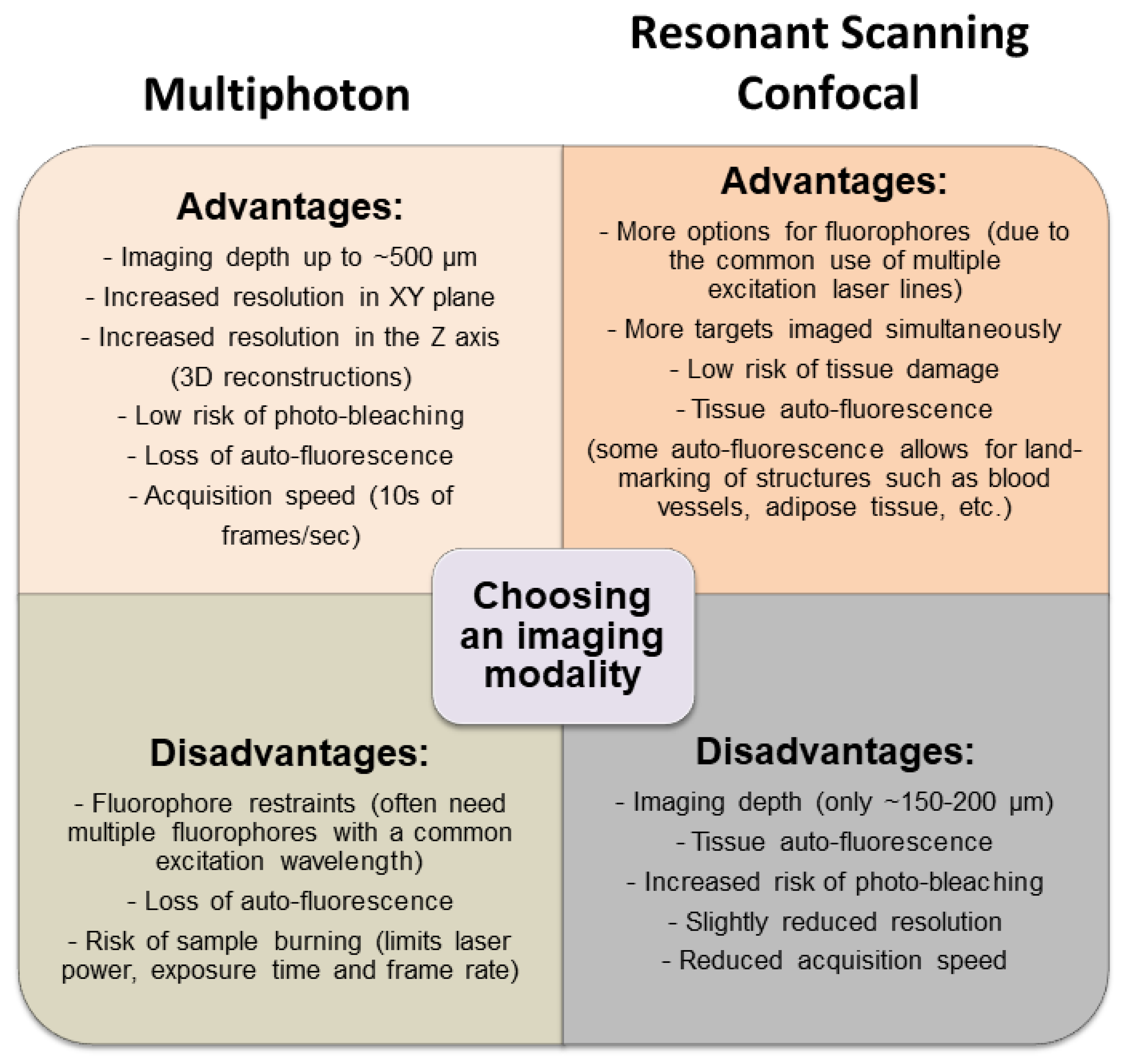
3.1. Multimodal IVM Imaging of the TME
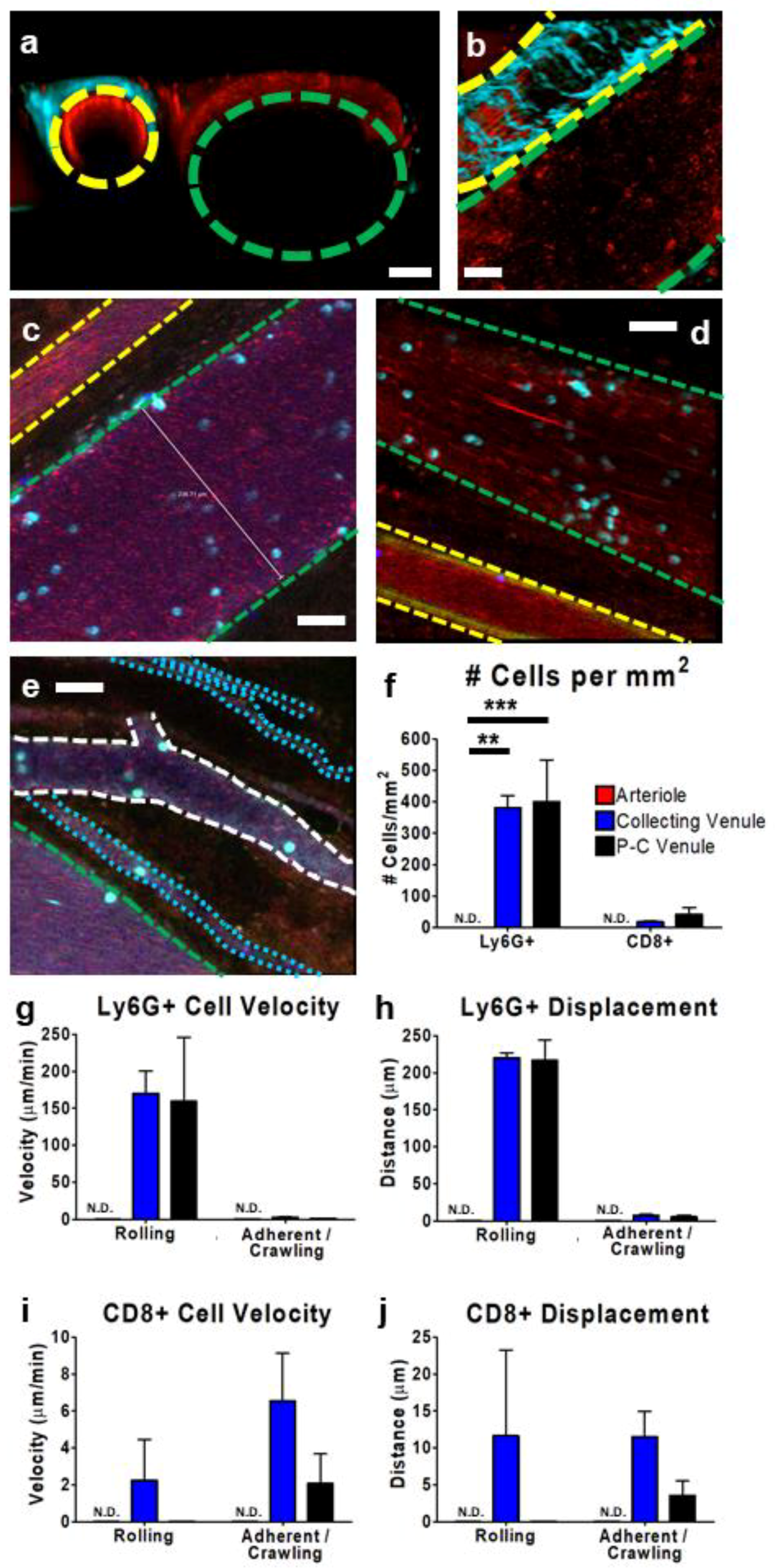
3.2. IVM of Leukocyte Behavior in Tumor Arterioles and Venules
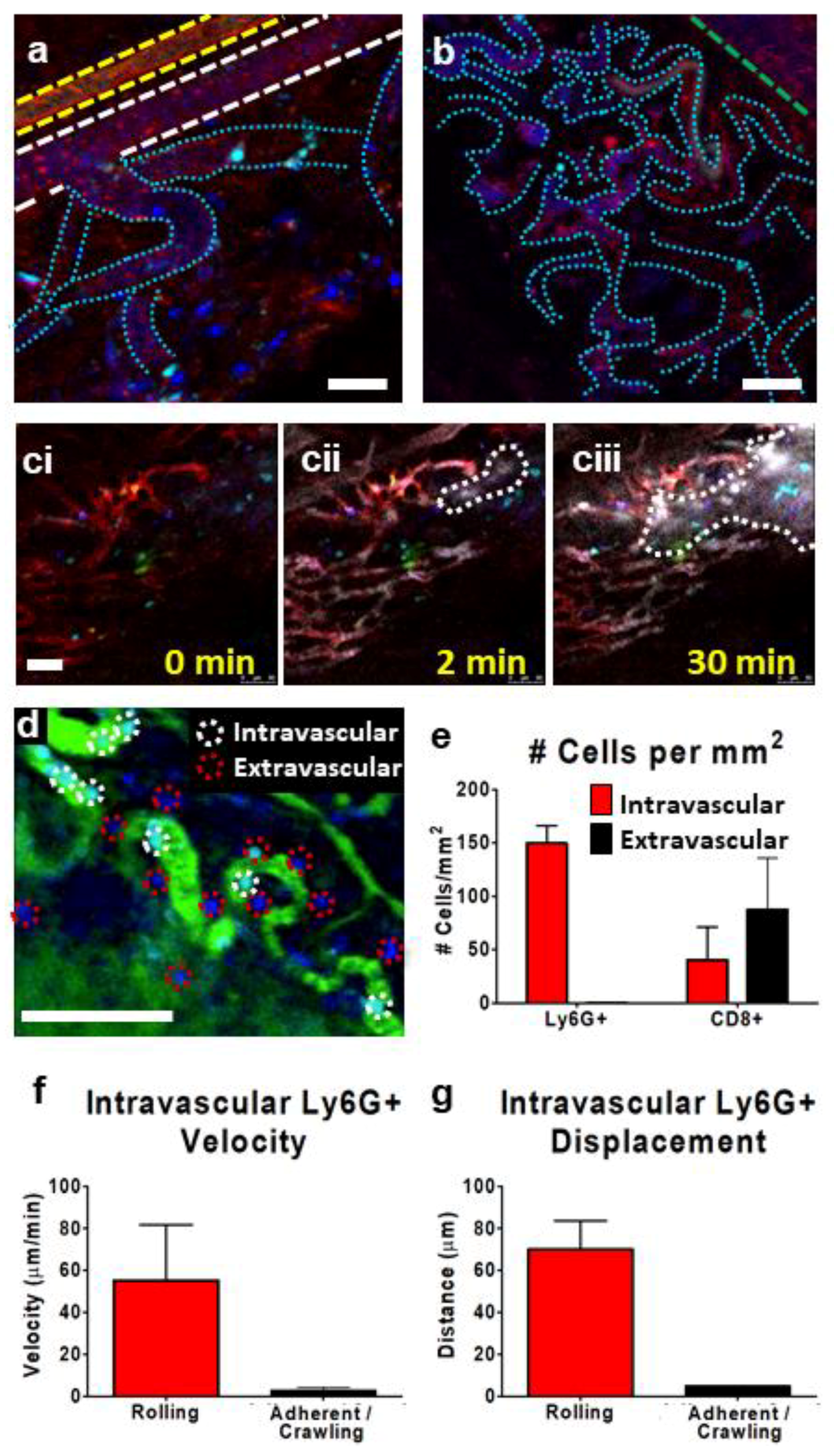
3.3. IVM of Leukocyte Behavior in Tumor Microcirculation/Capillaries
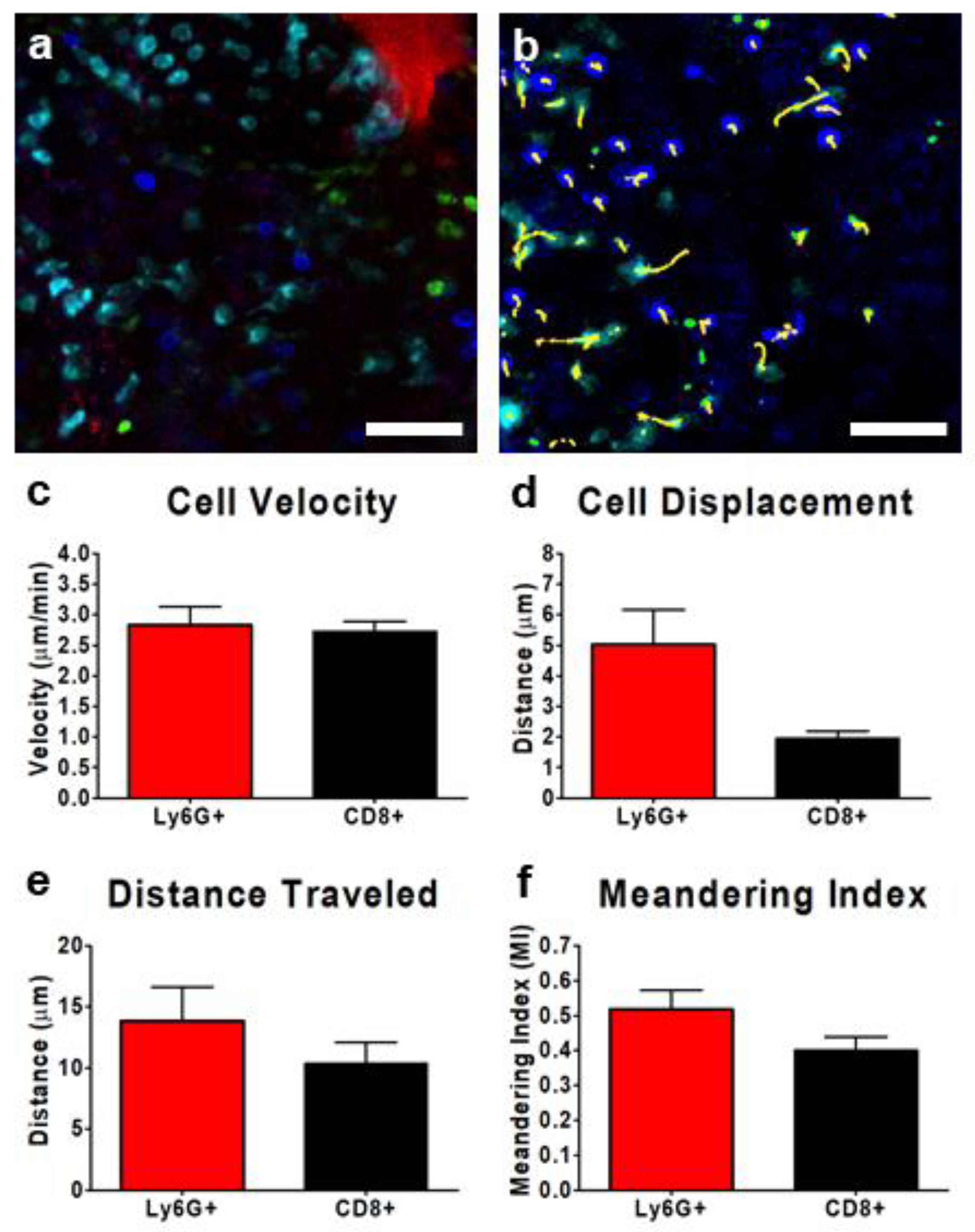
3.4. Tracking Leukocytes in the Tumor Interstitium by IVM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cohnheim, J. Untersuchungen über die Embolischen Process; August Hirschwald: Berlin, Germany, 1872. [Google Scholar]
- Waller, A. Microscopic observations on the perforation of the capillaries by the corpuscles of the blood, and on the origin of mucus and pus-globules. Lond. Edinb. Dublin Philos. Mag. 1846, 29, 397–405. [Google Scholar] [CrossRef][Green Version]
- Hall, M. A Critical and Experimental Essay on the Circulation of the Blood; R.B. Seeley and W. Burnside: London, UK, 1831. [Google Scholar]
- Johnston, B.; Burns, A.R.; Suematsu, M.; Issekutz, T.B.; Woodman, R.C.; Kubes, P. Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Investig. 1999, 103, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Hickey, M.J.; Murray, A.G.; Andonegui, G.; Kubes, P. Visualization of Plasmodium falciparum-endothelium interactions in human microvasculature: Mimicry of leukocyte recruitment. J. Exp. Med. 2000, 192, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Tang, H.L.; Cyster, J.G. Imaging of germinal center selection events during affinity maturation. Science 2007, 315, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Surewaard, B.G.; Wong, C.H.; Geoghegan, J.A.; Jenne, C.N.; Kubes, P. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 2016, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, C.; Schuchert, R.; Wagner, F.; Thaler, R.; Weinberger, T.; Pick, R.; Mass, E.; Ishikawa-Ankerhold, H.C.; Margraf, A.; Hutter, S.; et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat. Commun. 2018, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Perez-Camps, M.; Tian, J.; Chng, S.C.; Sem, K.P.; Sudhaharan, T.; Teh, C.; Wachsmuth, M.; Korzh, V.; Ahmed, S.; Reversade, B. Quantitative imaging reveals real-time Pou5f3-Nanog complexes driving dorsoventral mesendoderm patterning in zebrafish. eLife 2016, 5, e11475. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di, L.P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Christoffersson, G.; Vagesjo, E.; Vandooren, J.; Liden, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Green, J.A.; Gray, E.E.; Xu, Y.; Cyster, J.G. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat. Immunol. 2009, 10, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Lammermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmuller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.F.; McCachren, S.S., III; Lee, M.; Murphy, H.A.; Zhu, C.; Crouch, B.T.; Martin, H.L.; Erkanli, A.; Rajaram, N.; Ashcraft, K.A.; et al. Metaboloptics: Visualization of the tumor functional landscape via metabolic and vascular imaging. Sci. Rep. 2018, 8, 4171. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, W.; Sasai, K.; Akagi, T. Imaging Window Device for Subcutaneous Implantation Tumor. Methods Mol. Biol. 2018, 1763, 153–163. [Google Scholar] [PubMed]
- Seynhaeve, A.L.; Ten Hagen, T.L. High-Resolution Intravital Microscopy of Tumor Angiogenesis. Methods Mol. Biol. 2016, 1464, 115–127. [Google Scholar] [PubMed]
- Benjamin, D.C.; Hynes, R.O. Intravital imaging of metastasis in adult Zebrafish. BMC Cancer 2017, 17, 660. [Google Scholar] [CrossRef] [PubMed]
- Babes, L.; Kubes, P. Visualizing the Tumor Microenvironment of Liver Metastasis by Spinning Disk Confocal Microscopy. Methods Mol. Biol. 2016, 1458, 203–215. [Google Scholar] [PubMed]
- Weber, M.R.; Zuka, M.; Lorger, M.; Tschan, M.; Torbett, B.E.; Zijlstra, A.; Quigley, J.P.; Staflin, K.; Eliceiri, B.P.; Krueger, J.S.; et al. Activated tumor cell integrin alphavbeta3 cooperates with platelets to promote extravasation and metastasis from the blood stream. Thromb. Res. 2016, 140 (Suppl. 1), S27–S36. [Google Scholar] [CrossRef]
- Madsen, D.H.; Jurgensen, H.J.; Siersbaek, M.S.; Kuczek, D.E.; Grey, C.L.; Liu, S.; Behrendt, N.; Grontved, L.; Weigert, R.; Bugge, T.H. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017, 21, 3662–3671. [Google Scholar] [CrossRef] [PubMed]
- Belli, C.; Trapani, D.; Viale, G.; D’Amico, P.; Duso, B.A.; Della, V.P.; Orsi, F.; Curigliano, G. Targeting the microenvironment in solid tumors. Cancer Treat. Rev. 2018, 65, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Dzhandzhugazyan, K.N.; Guldberg, P.; Kirkin, A.F. Adoptive T cell cancer therapy. Nat. Mater. 2018, 17, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Stojdl, D.F.; Laird, G. Virus therapy for cancer. Sci. Am. 2014, 311, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Dastidar, H.; Zhang, C.; Zemp, F.J.; Lau, K.; Ernst, M.; Rakic, A.; Sikdar, S.; Rajwani, J.; Naumenko, V.; et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat. Commun. 2017, 8, 344. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.; Jenne, C.; Mahoney, D.J. Intravital Microscopy for Imaging the Tumor Microenvironment in Live Mice. Methods Mol. Biol. 2016, 1458, 217–230. [Google Scholar] [PubMed]
- Schindelin, J.; Rganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Parslow, A.; Cardona, A.; Bryson-Richardson, R.J. Sample drift correction following 4D confocal time-lapse imaging. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gailhouste, L.; Le, G.Y.; Odin, C.; Guyader, D.; Turlin, B.; Ezan, F.; Desille, Y.; Guilbert, T.; Bessard, A.; Fremin, C.; et al. Fibrillar collagen scoring by second harmonic microscopy: A new tool in the assessment of liver fibrosis. J. Hepatol. 2010, 52, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Braverman, I.M. The cutaneous microcirculation. J. Investig. Dermatol. Symp. Proc. 2000, 5, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 2014, 7, a016345. [Google Scholar] [CrossRef] [PubMed]
- Lemaster, K.A.; Farid, Z.; Brock, R.W.; Shrader, C.D.; Goldman, D.; Jackson, D.N.; Frisbee, J.C. Altered post-capillary and collecting venular reactivity in skeletal muscle with metabolic syndrome. J. Physiol. 2017, 595, 5159–5174. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Kanwar, S. Histamine induces leukocyte rolling in post-capillary venules. A P-selectin-mediated event. J. Immunol. 1994, 152, 3570–3577. [Google Scholar] [PubMed]
- Jung, U.; Ley, K. Mice lacking two or all three selectins demonstrate overlapping and distinct functions for each selectin. J. Immunol. 1999, 162, 6755–6762. [Google Scholar] [PubMed]
- Von Andrian, U.H.; Mackay, C.R. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 2000, 343, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Al, F.H.; Kang, J.H.; Hwang, S.R.; Sung, S.; Alam, M.M.; Sa, K.H.; Nam, E.J.; Byun, Y.R.; Kang, Y.M. Stepwise inhibition of T cell recruitment at post-capillary venules by orally active desulfated heparins in inflammatory arthritis. PLoS ONE 2017, 12, e0176110. [Google Scholar]
- Horie, Y.; Wolf, R.; Chervenak, R.P.; Jennings, S.R.; Granger, D.N. T-lymphocytes contribute to hepatic leukostasis and hypoxic stress induced by gut ischemia-reperfusion. Microcirculation 1999, 6, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.; Broermann, A.; Li, H.; Khandoga, A.G.; Zarbock, A.; Krombach, F.; Goerge, T.; Schneider, S.W.; Jones, C.; Nieswandt, B.; et al. Von Willebrand factor promotes leukocyte extravasation. Blood 2010, 116, 4712–4719. [Google Scholar] [CrossRef] [PubMed]
- Borlinghaus, R.T. Sensors and Measuring Techniques in Confocal Microscopy; Technological Readings; Leica Microsystems: Wetzlar, Germany, 2015; pp. 1–20. [Google Scholar]
- Kim, A.H.; Suleiman, H.; Shaw, A.S. New approaches in renal microscopy: Volumetric imaging and superresolution microscopy. Curr. Opin. Nephrol. Hypertens. 2016, 25, 159–167. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turk, M.; Naumenko, V.; Mahoney, D.J.; Jenne, C.N. Tracking Cell Recruitment and Behavior within the Tumor Microenvironment Using Advanced Intravital Imaging Approaches. Cells 2018, 7, 69. https://doi.org/10.3390/cells7070069
Turk M, Naumenko V, Mahoney DJ, Jenne CN. Tracking Cell Recruitment and Behavior within the Tumor Microenvironment Using Advanced Intravital Imaging Approaches. Cells. 2018; 7(7):69. https://doi.org/10.3390/cells7070069
Chicago/Turabian StyleTurk, Madison, Victor Naumenko, Douglas J. Mahoney, and Craig N. Jenne. 2018. "Tracking Cell Recruitment and Behavior within the Tumor Microenvironment Using Advanced Intravital Imaging Approaches" Cells 7, no. 7: 69. https://doi.org/10.3390/cells7070069
APA StyleTurk, M., Naumenko, V., Mahoney, D. J., & Jenne, C. N. (2018). Tracking Cell Recruitment and Behavior within the Tumor Microenvironment Using Advanced Intravital Imaging Approaches. Cells, 7(7), 69. https://doi.org/10.3390/cells7070069