Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte


Abstract
1. Introduction
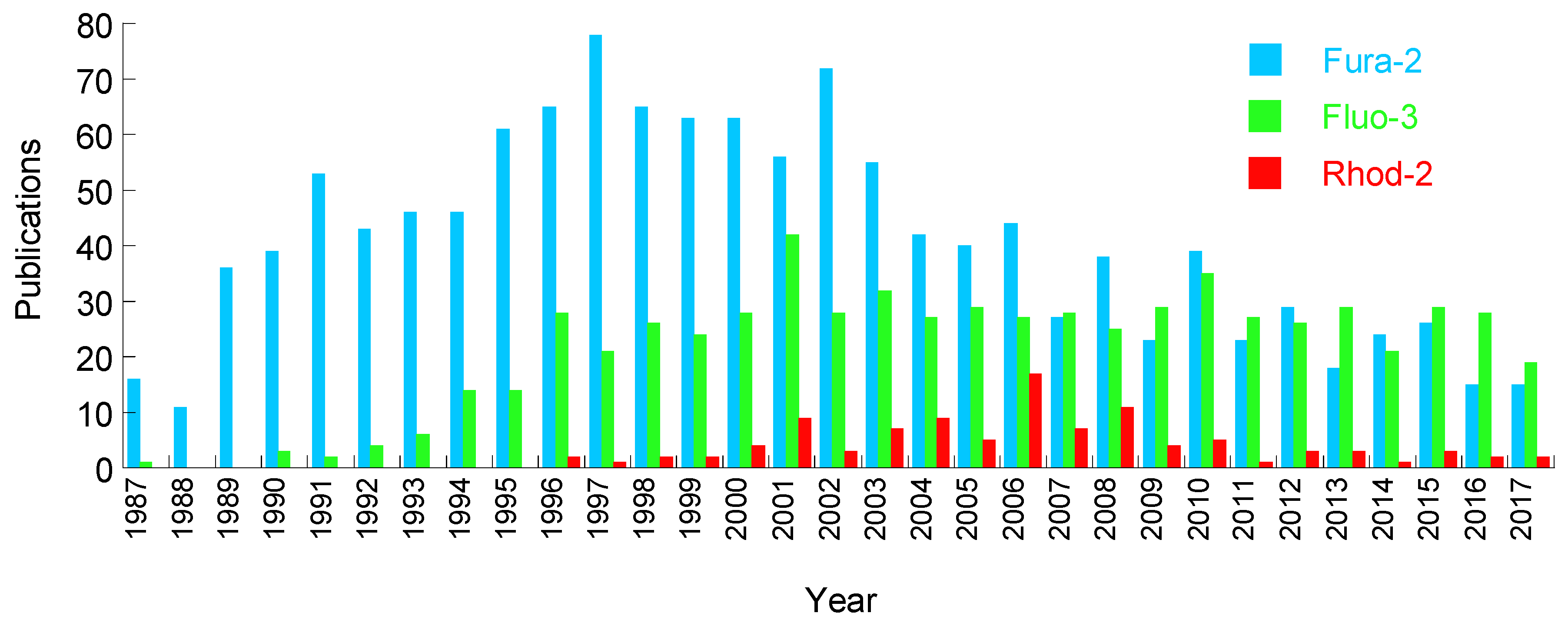
2. Chemical Dyes
2.1. Calcium Dyes
2.1.1. Calibration
2.1.2. Subcellular Applications
2.1.3. Disadvantages of the Calcium Dyes
2.2. Non-Calcium Sensitive Dyes
2.2.1. Sodium Indicators
2.2.2. Voltage Dyes
2.2.3. Others
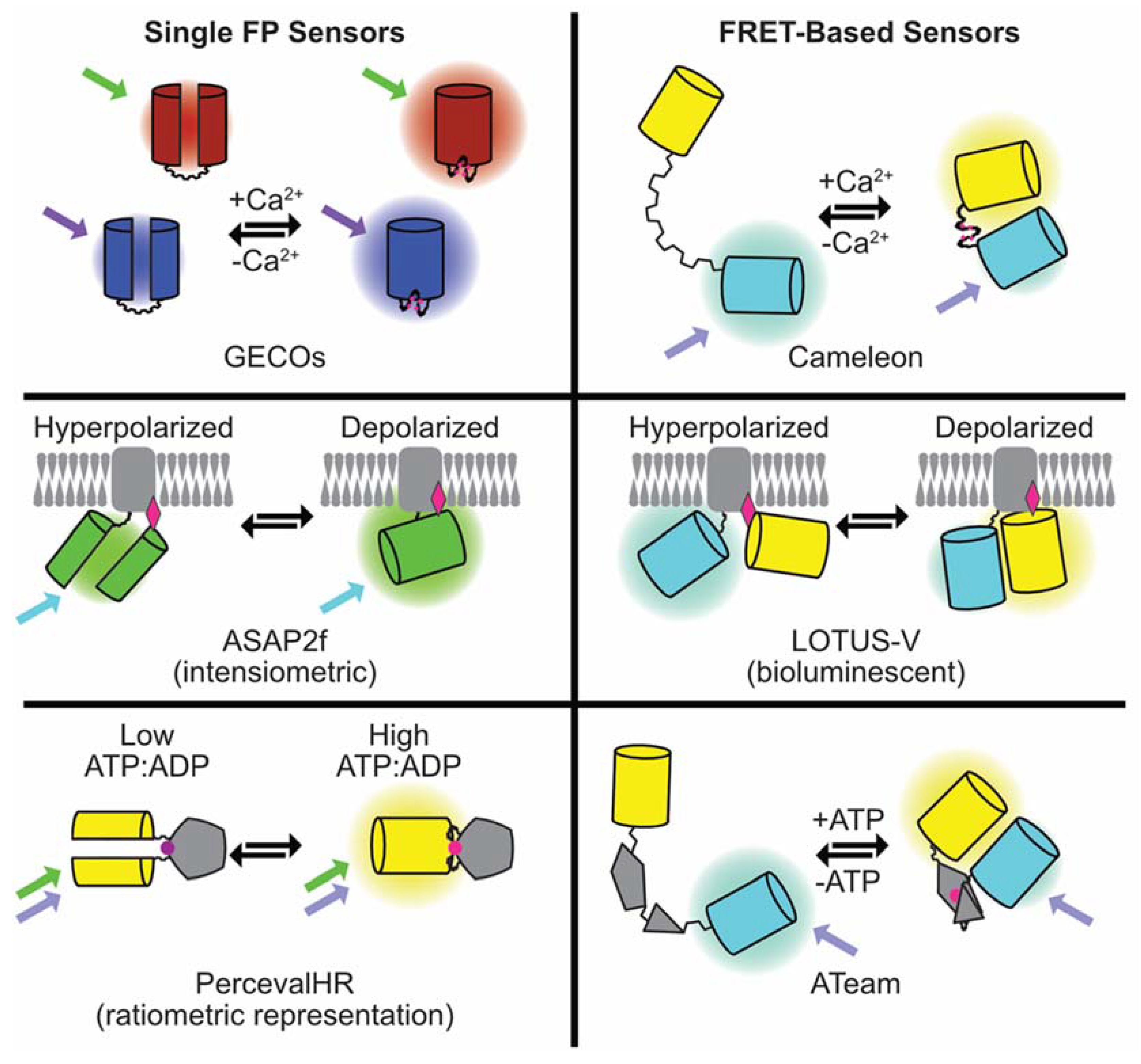
3. Genetically-Encoded Sensors
3.1. Genetically-Encoded Calcium Indicators
- (1)
- In head-to-head comparisons between GCaMP2, and Fura2, in the same cells, at the same time, the dye appears faster (on and off). Subsequent iterations have improved the speed of the GECI response.
- (2)
- When compared to Fura2, GCaMP3 produced a comparable signal to noise ratio, but with a Ca2+ buffering capacity at least an order of magnitude smaller than the dye.
- (3)
- The ratiometric indicator TN-XXL (Kd 0.8 μM, 136.5% signal change) [63] and the intensiometric indicator Flash Pericam (Kd 0.7 μM, 700% signal change) [61] were non-functional in adult cardiomyocytes, even though cells were successfully infected and in vitro performance suggested appropriate Kd and dynamic range. Therefore, it cannot be assumed that all potentially appropriate indicators will work in all cardiac model systems until they are tested. Since dyes change their Kd in the cellular environment [14] it is possible that protein-based indicators face similar challenges. This may make reliable and accurate calibration extremely difficult.
- (4)
- Although the reported dissociation rates for the GECIs in vitro at room temperature can be close to one second, in cellular preparations the observed decay (τOff) rates are adequate to see signals return to baseline with all indicators tested.
- (5)
- Adenovirally-delivered YC3.6 in a rat adult ventricular cardiomyocyte model at 48 h had an order of magnitude difference in the expression level (~0.4–4 μM), which corresponded to a similar variation in observed emission intensity.
3.1.1. Extending the Palette of GECIs; the GECOs
3.1.2. Affinity GECO Variants
3.2. GCaMP Series
3.3. RCAMPs
Undesirable Properties of the Red GECIs
3.4. Bioluminescent Calcium Indicators
3.5. Genetically-Encoded Voltage Indicators
3.6. Beyond Ca2+ and Voltage—Sensors for Cellular Energy (ATP/ADP) and Signaling Cascades (cAMP)
3.6.1. ATP and ATP/ADP Indicators
3.6.2. Cyclic Purine Metabolites cAMP and cGMP
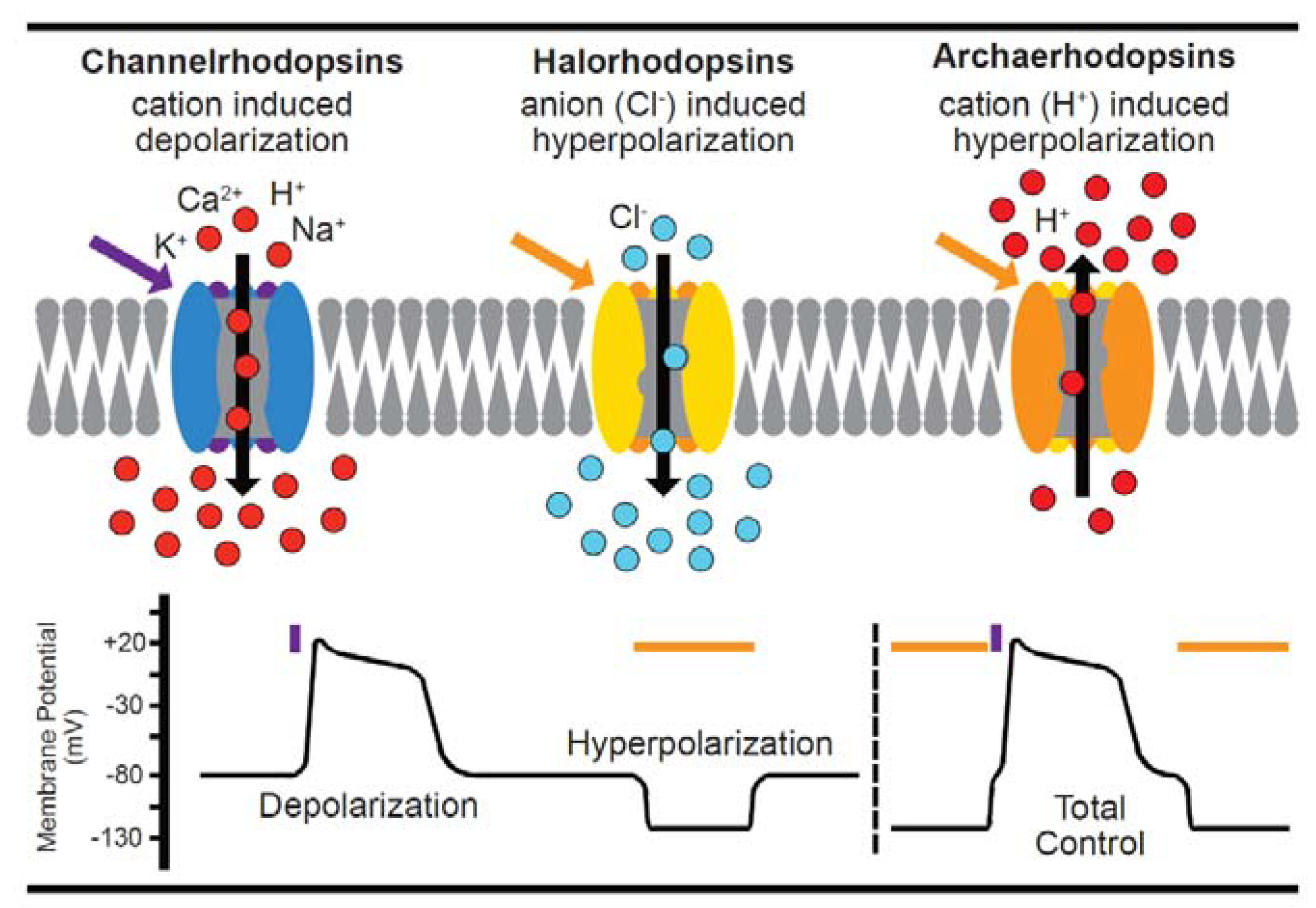
4. Genetically-Encoded Actuators (Optogenetics) in Single Cardiac Cells
Considerations for Optogenetic Application in Cardiomyocytes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ridgway, E.B.; Ashley, C.C. Calcium transients in single muscle fibers. Biochem. Biophys. Res. Commun. 1967, 29, 229–234. [Google Scholar] [CrossRef]
- Tsien, R.Y. New calcium indicators and buffers with high selectivity against magnesium and protons: Design, synthesis, and properties of prototype structures. Biochemistry 1980, 19, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.; Pozzan, T. Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 1989, 172, 230–262. [Google Scholar] [PubMed]
- Powell, T.; Tatham, P.E.; Twist, V.W. Cytoplasmic free calcium measured by quin2 fluorescence in isolated ventricular myocytes at rest and during potassium-depolarization. Biochem. Biophys. Res. Commun. 1984, 122, 1012–1020. [Google Scholar] [CrossRef]
- Sheu, S.S.; Sharma, V.K.; Banerjee, S.P. Measurement of cytosolic free calcium concentration in isolated rat ventricular myocytes with quin 2. Circ. Res. 1984, 55, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Minta, A.; Kao, J.P.; Tsien, R.Y. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989, 264, 8171–8178. [Google Scholar] [PubMed]
- Gee, K.R.; Archer, E.A.; Lapham, L.A.; Leonard, M.E.; Zhou, Z.L.; Bingham, J.; Diwu, Z. New ratiometric fluorescent calcium indicators with moderately attenuated binding affinities. Bioorg. Med. Chem. Lett. 2000, 10, 1515–1518. [Google Scholar] [CrossRef]
- Shannon, T.R.; Guo, T.; Bers, D.M. Ca2+ scraps: Local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003, 93, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Niggli, E.; Lederer, W.J. Real-time confocal microscopy and calcium measurements in heart muscle cells: Towards the development of a fluorescence microscope with high temporal and spatial resolution. Cell Calcium 1990, 11, 121–130. [Google Scholar] [CrossRef]
- Mackenzie, L.; Bootman, M.D.; Laine, M.; Berridge, M.J.; Thuring, J.; Holmes, A.; Li, W.H.; Lipp, P. The role of inositol 1,4,5-trisphosphate receptors in Ca(2+) signalling and the generation of arrhythmias in rat atrial myocytes. J. Physiol. 2002, 541, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys. J. 1995, 68, 1453–1460. [Google Scholar] [CrossRef]
- Cannell, M.B.; Berlin, J.R.; Lederer, W.J. Intracellular calcium in cardiac myocytes: Calcium transients measured using fluorescence imaging. Soc. Gen. Physiol. Ser. 1987, 42, 201–214. [Google Scholar] [PubMed]
- Thomas, D.; Tovey, S.C.; Collins, T.J.; Bootman, M.D.; Berridge, M.J.; Lipp, P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 2000, 28, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.R.; Konishi, M. Ca2+ transients in cardiac myocytes measured with high and low affinity Ca2+ indicators. Biophys. J. 1993, 65, 1632–1647. [Google Scholar] [CrossRef]
- Li, Q.; Altschuld, R.A.; Stokes, B.T. Quantitation of intracellular free calcium in single adult cardiomyocytes by fura-2 fluorescence microscopy: Calibration of fura-2 ratios. Biochem. Biophys. Res. Commun. 1987, 147, 120–126. [Google Scholar] [PubMed]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C.S. Hypertrophic cardiomyopathy mutations increase myofilament Ca(2+) buffering, alter intracellular Ca(2+) handling and stimulate Ca(2+) dependent signalling. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.M.; Etzler, J.C.; Watts, L.T.; Zheng, W.; Lechleiter, J.D. Chemical calcium indicators. Methods 2008, 46, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Trafford, A.W.; Diaz, M.E.; Eisner, D.A. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 1999, 437, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Schober, T.; Huke, S.; Venkataraman, R.; Gryshchenko, O.; Kryshtal, D.; Hwang, H.S.; Baudenbacher, F.J.; Knollmann, B.C. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res. 2012, 111, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Yan, P.; Loew, L.M.; Poggesi, C.; Cerbai, E.; Pavone, F.S.; Sacconi, L. T-Tubular Electrical Defects Contribute to Blunted beta-Adrenergic Response in Heart Failure. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Ferrantini, C.; Scardigli, M.; Coppini, R.; Mazzoni, L.; Lazzeri, E.; Pioner, J.M.; Scellini, B.; Guo, A.; Song, L.S.; et al. Novel insights on the relationship between T-tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 91, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Csordas, P.; Schneider, T.; Hajnoczky, G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol. 2000, 529 Pt 3, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Maack, C. Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 2010, 122, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, R.; Holcomb, M.R.; Harder, R.; Knollmann, B.C.; Baudenbacher, F. Ratiometric imaging of calcium during ischemia-reperfusion injury in isolated mouse hearts using Fura-2. Biomed. Eng. Online 2012, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Kress, B.T.; Lu, Y.; Chandler-Militello, D.; Benraiss, A.; Nedergaard, M. Fluorescent Ca(2+) indicators directly inhibit the Na,K-ATPase and disrupt cellular functions. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Sparrow, A.J.; Broyles, C.N.; Sievert, K.; Chang, Y.-F.; Brook, F.A.; Zhang, X.; Watkins, H.; Abassi, Y.A.; Geeves, M.A.; et al. Measurement of myofilament calcium in living cardiomyocytes using a targeted genetically encoded indicator. bioRxiv 2018. [Google Scholar] [CrossRef]
- Minta, A.; Tsien, R.Y. Fluorescent indicators for cytosolic sodium. J. Biol. Chem. 1989, 264, 19449–19457. [Google Scholar] [PubMed]
- Levi, A.J.; Lee, C.O.; Brooksby, P. Properties of the fluorescent sodium indicator “SBFI” in rat and rabbit cardiac myocytes. J. Cardiovasc. Electrophysiol. 1994, 5, 241–257. [Google Scholar] [PubMed]
- Donoso, P.; Mill, J.G.; O’Neill, S.C.; Eisner, D.A. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J. Physiol. 1992, 448, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Fluhler, E.; Burnham, V.G.; Loew, L.M. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry 1985, 24, 5749–5755. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Lin, J.Y.; Frady, E.P.; Steinbach, P.A.; Kristan, W.B., Jr.; Tsien, R.Y. Optically monitoring voltage in neurons by photo-induced electron transfer through molecular wires. Proc. Natl. Acad. Sci. USA 2012, 109, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Acker, C.D.; Zhou, W.L.; Lee, P.; Bollensdorff, C.; Negrean, A.; Lotti, J.; Sacconi, L.; Antic, S.D.; Kohl, P.; et al. Palette of fluorinated voltage-sensitive hemicyanine dyes. Proc. Natl. Acad. Sci. USA 2012, 109, 20443–20448. [Google Scholar] [CrossRef] [PubMed]
- Matiukas, A.; Mitrea, B.G.; Qin, M.; Pertsov, A.M.; Shvedko, A.G.; Warren, M.D.; Zaitsev, A.V.; Wuskell, J.P.; Wei, M.D.; Watras, J.; et al. Near-infrared voltage-sensitive fluorescent dyes optimized for optical mapping in blood-perfused myocardium. Heart Rhythm 2007, 4, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Knisley, S.B.; Blitchington, T.F.; Hill, B.C.; Grant, A.O.; Smith, W.M.; Pilkington, T.C.; Ideker, R.E. Optical measurements of transmembrane potential changes during electric field stimulation of ventricular cells. Circ. Res. 1993, 72, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.S.; Kaplan, D.T.; Kanai, A.; Jackson, L.; Garan, H.; Cohen, R.J.; Salama, G. Repolarization inhomogeneities in ventricular myocardium change dynamically with abrupt cycle length shortening. Circulation 1991, 84, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, L.; Ferrantini, C.; Lotti, J.; Coppini, R.; Yan, P.; Loew, L.M.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 5815–5819. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.; Figueroa, L.; Fitts, R.; Rios, E. Confocal imaging of transmembrane voltage by SEER of di-8-ANEPPS. J. Gen. Physiol. 2013, 141, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Streit, J.; Kleinlogel, S. Dynamic all-optical drug screening on cardiac voltage-gated ion channels. Sci. Rep. 2018, 8, 1153. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.E.; Tsien, R.Y. Improved indicators of cell membrane potential that use fluorescence resonance energy transfer. Chem. Biol. 1997, 4, 269–277. [Google Scholar] [CrossRef]
- DiFranco, M.; Capote, J.; Quinonez, M.; Vergara, J.L. Voltage-dependent dynamic FRET signals from the transverse tubules in mammalian skeletal muscle fibers. J. Gen. Physiol. 2007, 130, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Van Wagoner, D.R.; Mazgalev, T.N.; Tchou, P.J.; Efimov, I.R. Voltage-sensitive dye RH421 increases contractility of cardiac muscle. Can. J. Physiol. Pharmacol. 1998, 76, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Hayashi, S.; Ojima, A.; Taniguchi, T.; Miyamoto, N.; Nakamori, C.; Nagasawa, C.; Kitamura, T.; Osada, T.; Honda, Y.; et al. Improvement of acquisition and analysis methods in multi-electrode array experiments with iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2015, 75, 17–26. [Google Scholar] [CrossRef] [PubMed]
- McPheeters, M.T.; Wang, Y.T.; Werdich, A.A.; Jenkins, M.W.; Laurita, K.R. An infrared optical pacing system for screening cardiac electrophysiology in human cardiomyocytes. PLoS ONE 2017, 12, e0183761. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.F.; Lee, W.S.; Wang, I.C.; Chen, T.I.; Chen, T.L.; Yang, K.T. Intermittent hypoxia-generated ROS contributes to intracellular zinc regulation that limits ischemia/reperfusion injury in adult rat cardiomyocyte. J. Mol. Cell. Cardiol. 2018, 118, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, M.; Inoue, H.; Konishi, M. Physiological pathway of magnesium influx in rat ventricular myocytes. Biophys. J. 2014, 107, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.A.; Rabkin, S.W. Cytosolic free magnesium in cardiac myocytes: Identification of a Mg2+ influx pathway. Biochem. Biophys. Res. Commun. 1990, 167, 1406–1412. [Google Scholar] [CrossRef]
- Griffiths, E.J. Calcium handling and cell contraction in rat cardiomyocytes depleted of intracellular magnesium. Cardiovasc. Res. 2000, 47, 116–123. [Google Scholar] [CrossRef]
- Swietach, P.; Zaniboni, M.; Stewart, A.K.; Rossini, A.; Spitzer, K.W.; Vaughan-Jones, R.D. Modelling intracellular H(+) ion diffusion. Prog. Biophys. Mol. Biol. 2003, 83, 69–100. [Google Scholar] [CrossRef]
- Ashley, C.C.; Ridgway, E.B. On the relationships between membrane potential, calcium transient and tension in single barnacle muscle fibres. J. Physiol. 1970, 209, 105–130. [Google Scholar] [CrossRef] [PubMed]
- Tomosugi, W.; Matsuda, T.; Tani, T.; Nemoto, T.; Kotera, I.; Saito, K.; Horikawa, K.; Nagai, T. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods 2009, 6, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, L.; Matsuda, T.; Zhao, Y.; Rebane, A.; Drobizhev, M.; Chang, Y.F.; Araki, S.; Arai, Y.; March, K.; et al. Improved orange and red Ca(2)+/− indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 2013, 4, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Yao, A.; Oyama, T.; Hasegawa, H.; Akazawa, H.; Toko, H.; Nagai, T.; Kinugawa, K.; Kohmoto, O.; Maruyama, K.; et al. Direct measurement of Ca2+ concentration in the SR of living cardiac myocytes. Biochem. Biophys. Res. Commun. 2004, 314, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, L.; Scholz, A.; Tian, Q.; Ruppenthal, S.; Tabellion, W.; Wiesen, K.; Katus, H.A.; Muller, O.J.; Kotlikoff, M.I.; Lipp, P. Genetically encoded Ca2+ indicators in cardiac myocytes. Circ. Res. 2014, 114, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Mank, M.; Santos, A.F.; Direnberger, S.; Mrsic-Flogel, T.D.; Hofer, S.B.; Stein, V.; Hendel, T.; Reiff, D.F.; Levelt, C.; Borst, A.; et al. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat. Methods 2008, 5, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Kettlewell, S.; Cabrero, P.; Nicklin, S.A.; Dow, J.A.; Davies, S.; Smith, G.L. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J. Mol. Cell. Cardiol. 2009, 46, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Abdelfattah, A.S.; Miraucourt, L.S.; Kutsarova, E.; Ruangkittisakul, A.; Zhou, H.; Ballanyi, K.; Wicks, G.; Drobizhev, M.; Rebane, A.; et al. A long Stokes shift red fluorescent Ca2+ indicator protein for two-photon and ratiometric imaging. Nat. Commun. 2014, 5, 5262. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Prole, D.L.; Shen, Y.; Lin, Z.; Gnanasekaran, A.; Liu, Y.; Chen, L.; Zhou, H.; Chen, S.R.; Usachev, Y.M.; et al. Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem. J. 2014, 464, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Broyles, C.N.; Brook, F.A.; Davies, M.J.; Turtle, C.W.; Nagai, T.; Daniels, M.J. Non-invasive phenotyping and drug testing in single cardiomyocytes or beta-cells by calcium imaging and optogenetics. PLoS ONE 2017, 12, e0174181. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Dana, H.; Abdelfattah, A.S.; Patel, R.; Shea, J.; Molina, R.S.; Rawal, B.; Rancic, V.; Chang, Y.F.; Wu, L.; et al. A genetically encoded Ca(2+) indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Hires, S.A.; Mao, T.; Huber, D.; Chiappe, M.E.; Chalasani, S.H.; Petreanu, L.; Akerboom, J.; McKinney, S.A.; Schreiter, E.R.; et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 2009, 6, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Sasaki, T.; Sadakari, J.; Gengyo-Ando, K.; Kagawa-Nagamura, Y.; Kobayashi, C.; Ikegaya, Y.; Nakai, J. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. PLoS ONE 2012, 7, e51286. [Google Scholar] [CrossRef] [PubMed]
- Shiba, Y.; Fernandes, S.; Zhu, W.Z.; Filice, D.; Muskheli, V.; Kim, J.; Palpant, N.J.; Gantz, J.; Moyes, K.W.; Reinecke, H.; et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 2012, 489, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Maddah, M.; Heidmann, J.D.; Mandegar, M.A.; Walker, C.D.; Bolouki, S.; Conklin, B.R.; Loewke, K.E. A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Rep. 2015, 4, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Huebsch, N.; Loskill, P.; Mandegar, M.A.; Marks, N.C.; Sheehan, A.S.; Ma, Z.; Mathur, A.; Nguyen, T.N.; Yoo, J.C.; Judge, L.M.; et al. Automated Video-Based Analysis of Contractility and Calcium Flux in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Cultured over Different Spatial Scales. Tissue Eng. Part C Methods 2015, 21, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, G.T.; Chaudhary, K.W.; Atwater, N.; Nguyen, C.; Brown, B.S.; McNeish, J.D.; Cohen, A.E.; Kralj, J.M. Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. J. Pharmacol. Toxicol. Methods 2016, 81, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Werley, C.A.; Chien, M.P.; Gaublomme, J.; Shekhar, K.; Butty, V.; Yi, B.A.; Kralj, J.M.; Bloxham, W.; Boyer, L.A.; Regev, A.; et al. Geometry-dependent functional changes in iPSC-derived cardiomyocytes probed by functional imaging and RNA sequencing. PLoS ONE 2017, 12, e0172671. [Google Scholar] [CrossRef] [PubMed]
- Ai, M.; Mills, H.; Kanai, M.; Lai, J.; Deng, J.; Schreiter, E.; Looger, L.; Neubert, T.; Suh, G. Green-to-Red Photoconversion of GCaMP. PLoS ONE 2015, 10, e0138127. [Google Scholar] [CrossRef] [PubMed]
- Kredel, S.; Oswald, F.; Nienhaus, K.; Deuschle, K.; Rocker, C.; Wolff, M.; Heilker, R.; Nienhaus, G.U.; Wiedenmann, J. mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS ONE 2009, 4, e4391. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Carreras Calderon, N.; Tian, L.; Wabnig, S.; Prigge, M.; Tolo, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Sasaki, T.; Kobayashi, C.; Ikegaya, Y.; Nakai, J. An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS ONE 2012, 7, e39933. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Takeuchi, A.; Horigane, S.; Ohkura, M.; Gengyo-Ando, K.; Fujii, H.; Kamijo, S.; Takemoto-Kimura, S.; Kano, M.; Nakai, J.; et al. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat. Methods 2015, 12, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Chang, Y.F.; Horikawa, K.; Hatsugai, N.; Higuchi, Y.; Hashida, M.; Yoshida, Y.; Matsuda, T.; Arai, Y.; Nagai, T. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat. Commun. 2012, 3, 1262. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kimura, T.; Shinoda, H.; Bai, G.; Daniels, M.J.; Arai, Y.; Nakano, M.; Nagai, T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016, 7, 13718. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.N.; Suzuki, K.; Iwano, M.; Matsuda, T.; Nagai, T. Bioluminescent Low-Affinity Ca(2+) Indicator for ER with Multicolor Calcium Imaging in Single Living Cells. ACS Chem. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Mange, J.S.; Mills, R.W.; Macri, V.S.; Jang, M.Y.; Butte, F.N.; Ellinor, P.T.; Milan, D.J. Rapid cellular phenotyping of human pluripotent stem cell-derived cardiomyocytes using a genetically encoded fluorescent voltage sensor. Stem Cell Rep. 2014, 2, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zou, P.; Cohen, A.E. Voltage imaging with genetically encoded indicators. Curr. Opin. Chem. Biol. 2017, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Oberhofer, M.; Ruppenthal, S.; Scholz, A.; Buschmann, V.; Tsutsui, H.; Miyawaki, A.; Zeug, A.; Lipp, P.; Kaestner, L. Optical action potential screening on adult ventricular myocytes as an alternative QT-screen. Cell. Physiol. Biochem. 2011, 27, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, L.; Tian, Q.; Kaiser, E.; Xian, W.; Muller, A.; Oberhofer, M.; Ruppenthal, S.; Sinnecker, D.; Tsutsui, H.; Miyawaki, A.; et al. Genetically Encoded Voltage Indicators in Circulation Research. Int. J. Mol. Sci. 2015, 16, 21626–21642. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Tsutsui, H.; Suzuki, K.; Agetsuma, M.; Arai, Y.; Jinno, Y.; Bai, G.; Daniels, M.J.; Okamura, Y.; Matsuda, T.; et al. Genetically encoded bioluminescent voltage indicator for multi-purpose use in wide range of bioimaging. Sci. Rep. 2017, 7, 42398. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Geiller, T.; Jung, A.; Nakajima, R.; Song, Y.K.; Baker, B.J. Improving a genetically encoded voltage indicator by modifying the cytoplasmic charge composition. Sci. Rep. 2017, 7, 8286. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.S.; Farhi, S.L.; Zhao, Y.; Brinks, D.; Zou, P.; Ruangkittisakul, A.; Platisa, J.; Pieribone, V.A.; Ballanyi, K.; Cohen, A.E.; et al. A Bright and Fast Red Fluorescent Protein Voltage Indicator That Reports Neuronal Activity in Organotypic Brain Slices. J. Neurosci. 2016, 36, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; St-Pierre, F.; Sun, X.; Ding, X.; Lin, M.Z.; Clandinin, T.R. Subcellular Imaging of Voltage and Calcium Signals Reveals Neural Processing In Vivo. Cell 2016, 166, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell. Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.C.; Allshire, A.P.; Cobbold, P.H. Bioluminescent measurement in single cardiomyocytes of sudden cytosolic ATP depletion coincident with rigor. J. Mol. Cell. Cardiol. 1992, 24, 213–218. [Google Scholar] [CrossRef]
- Gribble, F.M.; Loussouarn, G.; Tucker, S.J.; Zhao, C.; Nichols, C.G.; Ashcroft, F.M. A novel method for measurement of submembrane ATP concentration. J. Biol. Chem. 2000, 275, 30046–30049. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, T.; Lippiat, J.D.; Ashcroft, F.M.; Rutter, G.A. ATP-dependent interaction of the cytosolic domains of the inwardly rectifying K+ channel Kir6.2 revealed by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2004, 101, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Nhat, K.P.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kakizuka, A.; Imamura, H. BTeam, a Novel BRET-based Biosensor for the Accurate Quantification of ATP Concentration within Living Cells. Sci. Rep. 2016, 6, 39618. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014, 4, 6522. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Hung, Y.P.; Yellen, G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat. Methods 2009, 6, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Tantama, M.; Martinez-Francois, J.R.; Mongeon, R.; Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun. 2013, 4, 2550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Wu, D.; Huang, Z.; Hou, T.; Jian, C.; Yu, P.; Lu, F.; Zhang, R.; Sun, T.; et al. Mitochondrial flashes regulate ATP homeostasis in the heart. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef] [PubMed]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Harada, K.; Ito, M.; Takizawa, M.; Wongso, D.; Tsuboi, T.; Kitaguchi, T. Generation of a cGMP Indicator with an Expanded Dynamic Range by Optimization of Amino Acid Linkers between a Fluorescent Protein and PDE5alpha. ACS Sens. 2017, 2, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol. J. 2014, 9, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Bruegmann, T.; Malan, D.; Hesse, M.; Beiert, T.; Fuegemann, C.J.; Fleischmann, B.K.; Sasse, P. Optogenetic control of heart muscle in vitro and in vivo. Nat. Methods 2010, 7, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Gepstein, L. Optogenetics for in vivo cardiac pacing and resynchronization therapies. Nat. Biotechnol. 2015, 33, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Klimas, A.; Ambrosi, C.M.; Yu, J.; Williams, J.C.; Bien, H.; Entcheva, E. OptoDyCE as an automated system for high-throughput all-optical dynamic cardiac electrophysiology. Nat. Commun. 2016, 7, 11542. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, H.T.; Zhou, Y.M.; Wang, X.; Wang, L.; Liu, Z.Q. Cardiac optogenetics: A novel approach to cardiovascular disease therapy. Europace 2017. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, C.M.; Boyle, P.M.; Chen, K.; Trayanova, N.A.; Entcheva, E. Optogenetics-enabled assessment of viral gene and cell therapy for restoration of cardiac excitability. Sci. Rep. 2015, 5, 17350. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Valiunas, V.; Lu, Z.; Bien, H.; Liu, H.; Wang, H.Z.; Rosati, B.; Brink, P.R.; Cohen, I.S.; Entcheva, E. Stimulating cardiac muscle by light: Cardiac optogenetics by cell delivery. Circ. Arrhythm. Electrophysiol. 2011, 4, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bruegmann, T.; Boyle, P.M.; Vogt, C.C.; Karathanos, T.V.; Arevalo, H.J.; Fleischmann, B.K.; Trayanova, N.A.; Sasse, P. Optogenetic defibrillation terminates ventricular arrhythmia in mouse hearts and human simulations. J. Clin. Investig. 2016, 126, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Kleinlogel, S.; Feldbauer, K.; Dempski, R.E.; Fotis, H.; Wood, P.G.; Bamann, C.; Bamberg, E. Ultra light-sensitive and fast neuronal activation with the Ca2+-permeable channelrhodopsin CatCh. Nat. Neurosci. 2011, 14, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bingen, B.O.; Engels, M.C.; Schalij, M.J.; Jangsangthong, W.; Neshati, Z.; Feola, I.; Ypey, D.L.; Askar, S.F.; Panfilov, A.V.; Pijnappels, D.A.; et al. Light-induced termination of spiral wave arrhythmias by optogenetic engineering of atrial cardiomyocytes. Cardiovasc. Res. 2014, 104, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Chater, T.E.; Henley, J.M.; Brown, J.T.; Randall, A.D. Voltage- and temperature-dependent gating of heterologously expressed channelrhodopsin-2. J. Neurosci. Methods 2010, 193, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Arai, Y.; Nagai, T. Optogenetic activation during detector “dead time” enables compatible real-time fluorescence imaging. Neurosci. Res. 2012, 73, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.T.; Rodriguez-Prados, M.; Navas-Navarro, P.; Rojo-Ruiz, J.; Garcia-Sancho, J. Using aequorin probes to measure Ca(2+) in intracellular organelles. Cell Calcium 2017, 64, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, T.M.; Safi, F.; Axelsson, A.S.; Mollet, I.G.; Rosengren, A.H. Optogenetic control of insulin secretion in intact pancreatic islets with beta-cell-specific expression of Channelrhodopsin-2. Islets 2014, 6, e28095. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Kanaporis, G.; Valiuniene, L.; Gordon, C.; Wang, H.Z.; Li, L.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; Brink, P.R. Coupling an HCN2-expressing cell to a myocyte creates a two-cell pacing unit. J. Physiol. 2009, 587, 5211–5226. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, K.; Lucero, R.V.; Ambrosi, C.M.; Entcheva, E. Cardiac Optogenetics: Enhancement by All-trans-Retinal. Sci. Rep. 2015, 5, 16542. [Google Scholar] [CrossRef] [PubMed]
- Entcheva, E. Cardiac optogenetics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1179–H1191. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Xu, J.; Lu, Z.; Klimas, A.; Chen, X.; Ambrosi, C.M.; Cohen, I.S.; Entcheva, E. Computational optogenetics: Empirically-derived voltage- and light-sensitive channelrhodopsin-2 model. PLoS Comput. Biol. 2013, 9, e1003220. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Indicator | Structure | Ca 2+ Kd (nM) | Excitation/Emission | Reference |
| BAPTA |  | 160 | Non-fluorescent | [2] |
| FLUO-3 |  | 325 | Ex 506 nmc Em 526 nm | [6] |
| FLUO-4 |  | 345 | Ex 494 nm Em 516 nm | [7] |
| Indicator | Structure | Ca2+ Kd (nM) | Excitation/Emission | Reference |
| FLUO-5-N |  | 135 × 103 | Ex 491 nm Em 516 nm | [8] |
| FURA-2 |  | 145 | Ex 340/380 nm Em 510 nm | [9] |
| INDO-1 |  | 230 | Ex 388 nm Em 400/475 nm | [9] |
| QUIN-2 |  | 115 | Ex 332 nm Em 493 nm | [3] |
| RHOD-2 |  | 370 | Ex 553 nm Em 576 nm | [6] |
| Indicator | Structure | Excitation:Emission | Reference |
| SBFI (Na+) |  | Ex 340/380 nm Em 510 nm | [31] |
| di-4-ANNEPS (voltage) |  | Ex 496 nm Em 705 nm | [34] |
| di-4-AN (F)EPPTEA (voltage) |  | Ex 444 nm Em 610 nm | [36] |
| di-4-AN BDQPQ (voltage) |  | Ex 488 nm Em 680 nm | [37] |
| FLUOVOLT (voltage) VF2.4.Cl |  | Ex 520 nm Em 551 nm | [35] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broyles, C.N.; Robinson, P.; Daniels, M.J. Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte. Cells 2018, 7, 51. https://doi.org/10.3390/cells7060051
Broyles CN, Robinson P, Daniels MJ. Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte. Cells. 2018; 7(6):51. https://doi.org/10.3390/cells7060051
Chicago/Turabian StyleBroyles, Connor N., Paul Robinson, and Matthew J. Daniels. 2018. "Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte" Cells 7, no. 6: 51. https://doi.org/10.3390/cells7060051
APA StyleBroyles, C. N., Robinson, P., & Daniels, M. J. (2018). Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte. Cells, 7(6), 51. https://doi.org/10.3390/cells7060051