Human-Induced Pluripotent Stem Cell Technology and Cardiomyocyte Generation: Progress and Clinical Applications

 ,
,  , and
, and 
Abstract
1. Introduction
2. Regulatory Pathways and Epigenetic Control of Cardiomyogenesis
2.1. Mesoderm Induction, Cardiac Specification, and Differentiation
2.2. The Wnt Signaling
2.3. Epigenetic Regulation of Human Cardiac Differentiation
2.4. The “Epigenetic Memory” in hiPSCs Differentiation Potential
3. Generation of CMs from hiPSCs Culture
3.1. EB Formation-Based Differentiation Protocol
3.2. Monolayer Culture-Based Differentiation Protocol
3.3. Large-Scale CM Differentiation in Suspension Culture
4. Morphological and Functional Properties of hiPSCs Derived CMs
5. hiPSC Paracrine Effects for Cardiac Repair and Regeneration
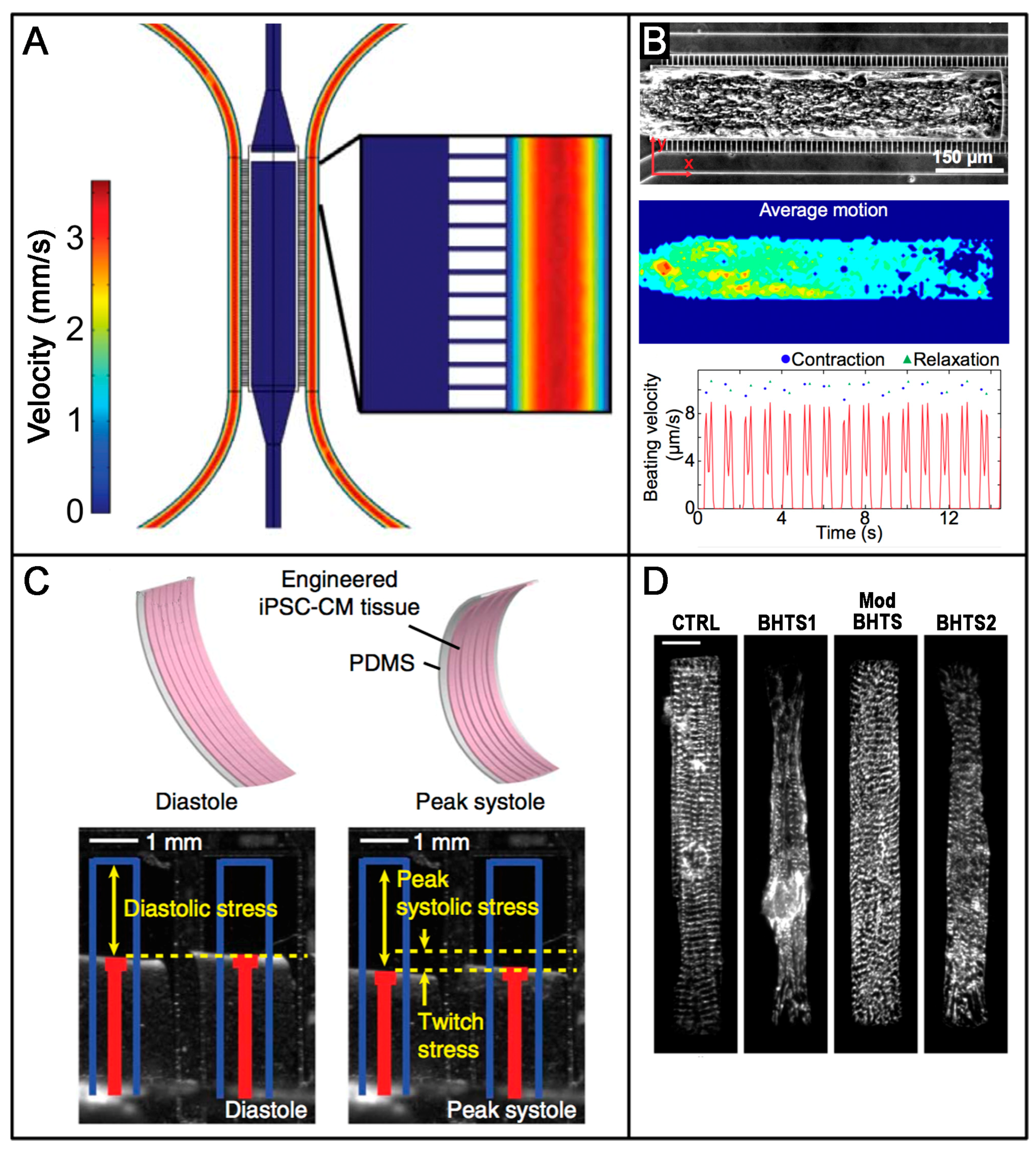
6. Advanced Technologies and Tissue Engineering: Novel Approaches for Studying hiPSC-Derived Cardiac Tissues
7. The Promise of hiPSCs-CMs in Biomedical Applications
8. The Immunological Challenges of hiPSCs and the Generation of Haplobank for the Cell Therapy
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart J. 2014, 35, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, M.; Ameri, P.; Marone, G.; Petretta, M.; Abete, P.; Di Lisa, F.; De Placido, S.; Bonaduce, D.; Tocchetti, C.G. Recent Advances on Pathophysiology, Diagnostic and Therapeutic Insights in Cardiac Dysfunction Induced by Antineoplastic Drugs. BioMed Res. Int. 2015, 2015, 138148. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Amabile, G.; Meissner, A. Induced pluripotent stem cells: Current progress and potential for regenerative medicine. Trends Mol. Med. 2009, 15, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lalit, P.A.; Hei, D.J.; Raval, A.N.; Kamp, T.J. Induced pluripotent stem cells for post-myocardial infarction repair: Remarkable opportunities and challenges. Circ. Res. 2014, 114, 1328–1345. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Hashizume, R.; Yoshizumi, T.; Blakney, A.K.; Ma, Z.; Wagner, W.R. Intramyocardial injection of a synthetic hydrogel with delivery of bFGF and IGF1 in a rat model of ischemic cardiomyopathy. Biomacromolecules 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Keller, G.; Gold, J.D.; Wu, J.C. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 2012, 10, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Mummery, C. Differentiation of Human Embryonic Stem Cells to Cardiomyocytes: Role of Coculture with Visceral Endoderm-Like Cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Costello, I.; Pimeisl, I.-M.; Dräger, S.; Bikoff, E.K.; Robertson, E.J.; Arnold, S.J. The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nat. Cell Biol. 2011, 13, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Jarsch, V.B.; Schwarz, F.; Nathan, P.; Gegg, M.; Lickert, H.; Franz, W.-M. Induction of MesP1 by Brachyury(T) generates the common multipotent cardiovascular stem cell. Cardiovasc. Res. 2011, 92, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. Development. The path to the heart and the road not taken. Science 2001, 291, 2327–2328. [Google Scholar] [CrossRef] [PubMed]
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. TEM 2012, 23, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. 2012, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A Temporal Chromatin Signature in Human Embryonic Stem Cells Identifies Regulators of Cardiac Development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.B.; Jones, T.A.; Herron, T.J.; Patel, S.R.; Day, S.M.; Noujaim, S.F.; Milstein, M.L.; Klos, M.; Furspan, P.B.; Jalife, J.; et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J. Clin. Investig. 2011, 121, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, J.D.; Riggs, A.D. An epigenetic perspective on the failing heart and pluripotent-derived-cardiomyocytes for cell replacement therapy. Front. Biol. 2015, 10, 11–27. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W.; Lee, S.-K. UTX, a Histone H3-Lysine 27 Demethylase, Acts as a Critical Switch to Activate the Cardiac Developmental Program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Sharma, A.; Wu, J.C. Genetic and Epigenetic Regulation of Human Cardiac Reprogramming and Differentiation in Regenerative Medicine. Annu. Rev. Genet. 2015, 49, 461–484. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, J.D.; Jung, M.; Chen, C.; Lin, Z.; Ye, J.; Godatha, S.; Lizhar, E.; Wu, X.; Hsu, D.; Couture, L.A.; et al. Mapping Human Pluripotent-to-Cardiomyocyte Differentiation: Methylomes, Transcriptomes, and Exon DNA Methylation “Memories”. EBioMedicine 2016, 4, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Dimmeler, S. Epigenetic regulation of cardiovascular differentiation. Cardiovasc. Res. 2011, 90, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Matkovich, S.J.; Edwards, J.R.; Grossenheider, T.C.; de Guzman Strong, C.; Dorn, G.W. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 12264–12269. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a Long Noncoding RNA Required for Cardiovascular Lineage Commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Long, H.; Zhou, C.; Zheng, S.; Wu, H.; Guo, T.; Wu, Q.; Zhong, T.; Wang, T. Long noncoding RNA Braveheart promotes cardiogenic differentiation of mesenchymal stem cells in vitro. Stem Cell Res. Ther. 2017, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The Tissue-Specific lncRNA Fendrr Is an Essential Regulator of Heart and Body Wall Development in the Mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.G.; van Mil, A.; van Vliet, P.; Metz, C.H.G.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 Regulate Differentiation and Proliferation in Human-Derived Cardiomyocyte Progenitor Cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, C.; Wang, Y.; Wang, W.; Wang, K.; Wu, X.; Li, Z.; Zhao, C.; Li, L.; Peng, L. Impact of miR-26b on cardiomyocyte differentiation in P19 cells through regulating canonical/non-canonical Wnt signalling. Cell Prolif. 2017, 50, e12371. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, N.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Isomi, M.; Nakashima, H.; Akiyama, M.; Wada, R.; Inagawa, K.; et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014, 33, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Soibam, B.; Benham, A.; Xu, X.; Chopra, M.; Peng, X.; Yu, W.; Bao, W.; Liang, R.; Azares, A.; et al. miR-322/-503 cluster is expressed in the earliest cardiac progenitor cells and drives cardiomyocyte specification. Proc. Natl. Acad. Sci. USA 2016, 113, 9551–9556. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Pan, B.; Zhou, H.; Liu, L.; Lv, T.; Zhu, J.; Huang, X.; Tian, J. Differentiation of mesenchymal stem cells into cardiomyocytes is regulated by miRNA-1-2 via WNT signaling pathway. J. Biomed. Sci. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Malizia, A.P.; Wang, D.-Z. MicroRNAs in cardiomyocyte development: MiRNA in cardiomyocyte development. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, K.T.; Jones, D.C.; Sperber, H.; Madan, A.; Fischer, K.A.; Rodriguez, M.L.; Pabon, L.; Zhu, W.-Z.; Tulloch, N.L.; Yang, X.; et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E2785–E2794. [Google Scholar] [CrossRef] [PubMed]
- Takaya, T.; Kawamura, T.; Morimoto, T.; Ono, K.; Kita, T.; Shimatsu, A.; Hasegawa, K. Identification of p300-targeted Acetylated Residues in GATA4 during Hypertrophic Responses in Cardiac Myocytes. J. Biol. Chem. 2008, 283, 9828–9835. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.M.; Margulies, K.B. Epigenetic Memory and Cardiac Cell Therapy∗. J. Am. Coll. Cardiol. 2014, 64, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Amabile, G.; Welner, R.S.; Nombela-Arrieta, C.; D’Alise, A.M.; Di Ruscio, A.; Ebralidze, A.K.; Kraytsberg, Y.; Ye, M.; Kocher, O.; Neuberg, D.S.; et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013, 121, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Freire, V.; Lee, A.S.; Hu, S.; Abilez, O.J.; Liang, P.; Lan, F.; Huber, B.C.; Ong, S.-G.; Hong, W.X.; Huang, M.; et al. Effect of Human Donor Cell Source on Differentiation and Function of Cardiac Induced Pluripotent Stem Cells. J. Am. Coll. Cardiol. 2014, 64, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Sirabella, D.; Cimetta, E.; Vunjak-Novakovic, G. “The state of the heart”: Recent advances in engineering human cardiac tissue from pluripotent stem cells. Exp. Biol. Med. 2015, 240, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Talkhabi, M.; Aghdami, N.; Baharvand, H. Human cardiomyocyte generation from pluripotent stem cells: A state-of-art. Life Sci. 2016, 145, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Senyei, G.D.; Hansen, J.; Kong, C.-W.; Azeloglu, E.U.; Stillitano, F.; Lieu, D.K.; Wang, J.; Ren, L.; Hulot, J.-S.; et al. Small molecule-mediated directed differentiation of human embryonic stem cells toward ventricular cardiomyocytes. Stem Cells Transl. Med. 2014, 3, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Braam, S.R.; Koutsis, K.; Ng, E.S.; Jenny, R.; Lagerqvist, E.L.; Biben, C.; Hatzistavrou, T.; Hirst, C.E.; Yu, Q.C.; et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods 2011, 8, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Schulte, J.S.; Heinick, A.; Piccini, I.; Rao, J.; Quaranta, R.; Zeuschner, D.; Malan, D.; Kim, K.-P.; Röpke, A.; et al. Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: Implications for in vitro maturation. Stem Cells 2015, 33, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: The matrix sandwich method. Circ. Res. 2012, 111, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Bao, X.; Zilberter, M.; Westman, M.; Fisahn, A.; Hsiao, C.; Hazeltine, L.B.; Dunn, K.K.; Kamp, T.J.; Palecek, S.P. Chemically defined, albumin-free human cardiomyocyte generation. Nat. Methods 2015, 12, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell–Derived CardiomyocytesNovelty and Significance. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.; Chen, A.; Reuveny, S.; Oh, S. An intermittent rocking platform for integrated expansion and differentiation of human pluripotent stem cells to cardiomyocytes in suspended microcarrier cultures. Stem Cell Res. 2014, 13, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.K.-L.; Chen, X.; Choo, A.B.H.; Reuveny, S.; Oh, S.K.W. Critical microcarrier properties affecting the expansion of undifferentiated human embryonic stem cells. Stem Cell Res. 2011, 7, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling Expansion and Cardiomyogenic Differentiation of Human Pluripotent Stem Cells in Scalable Suspension Culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes from Pluripotent Stem Cells: Scalable Production of hPSC-Derived Cardiomyocytes. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.A.; Ghinassi, B.; Izzicupo, P.; Di Ruscio, A.; Di Baldassarre, A. IL-6 Activates PI3K and PKCζ Signaling and Determines Cardiac Differentiation in Rat Embryonic H9c2 Cells. J. Cell. Physiol. 2016, 231, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [PubMed]
- Burridge, P.W.; Anderson, D.; Priddle, H.; Barbadillo Muñoz, M.D.; Chamberlain, S.; Allegrucci, C.; Young, L.E.; Denning, C. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells 2007, 25, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Mohr, J.C.; Zhang, J.; Azarin, S.M.; Soerens, A.G.; de Pablo, J.J.; Thomson, J.A.; Lyons, G.E.; Palecek, S.P.; Kamp, T.J. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 2010, 31, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Liang, H.; Huang, J.; Wang, J.; Chen, Y.; Chen, Z.; Yang, H.-T. Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 2013, 23, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Niebruegge, S.; Bauwens, C.L.; Peerani, R.; Thavandiran, N.; Masse, S.; Sevaptisidis, E.; Nanthakumar, K.; Woodhouse, K.; Husain, M.; Kumacheva, E.; et al. Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol. Bioeng. 2009, 102, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Parikh, A.; Tzanakakis, E.S. Cardiac Cell Generation from Encapsulated Embryonic Stem Cells in Static and Scalable Culture Systems. Cell Transplant. 2010, 19, 1397–1412. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Brito, C.; Sousa, M.F.Q.; Jensen, J.; Tostões, R.; Clemente, J.; Strehl, R.; Hyllner, J.; Carrondo, M.J.T.; Alves, P.M. Improving expansion of pluripotent human embryonic stem cells in perfused bioreactors through oxygen control. J. Biotechnol. 2010, 148, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Jawad, H.; Lyon, A.R.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Myocardial tissue engineering. Br. Med. Bull. 2008, 87, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Darkins, C.L.; Mandenius, C.-F. Design of large-scale manufacturing of induced pluripotent stem cell derived cardiomyocytes. Chem. Eng. Res. Des. 2014, 92, 1142–1152. [Google Scholar] [CrossRef]
- Lundy, S.D.; Zhu, W.-Z.; Regnier, M.; Laflamme, M.A. Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.; Tran, D.D.; George, S.C. Concise Review: Maturation Phases of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells 2013, 31, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence: Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, T.C.; Laflamme, M.A.; Murry, C.E. Proliferation of cardiomyocytes derived from human embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway. J. Mol. Cell. Cardiol. 2005, 39, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Johkura, K.; Takei, S.; Ogiwara, N.; Sasaki, K. Structural differentiation, proliferation, and association of human embryonic stem cell-derived cardiomyocytes in vitro and in their extracardiac tissues. J. Struct. Biol. 2007, 158, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Horigome, H.; Takahashi, M.I.; Asaka, M.; Shigemitsu, S.; Kandori, A.; Tsukada, K. Magnetocardiographic determination of the developmental changes in PQ, QRS and QT intervals in the foetus. Acta Paediatr. 2000, 89, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.A.; Klimanskaya, I.; McMahon, J.; Atienza, J.; Witmyer, J.; Zucker, J.P.; Wang, S.; Morton, C.C.; McMahon, A.P.; Powers, D.; et al. Derivation of Embryonic Stem-Cell Lines from Human Blastocysts. N. Engl. J. Med. 2004, 350, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Gherghiceanu, M.; Barad, L.; Novak, A.; Reiter, I.; Itskovitz-Eldor, J.; Binah, O.; Popescu, L.M. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell. Mol. Med. 2011, 15, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, C.L.; Peerani, R.; Niebruegge, S.; Woodhouse, K.A.; Kumacheva, E.; Husain, M.; Zandstra, P.W. Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells 2008, 26, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M.; Kellerman, S.E.; Moore, J.A.; Muffly, K.E.; Clark, L.C.; Reaves, P.Y.; Malec, K.B.; McKeown, P.P.; Schocken, D.D. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 1992, 86, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef] [PubMed]
- Izzicupo, P.; Ghinassi, B.; D’Amico, M.A.; Di Blasio, A.; Gesi, M.; Napolitano, G.; Gallina, S.; Di Baldassarre, A. Effects of ACE I/D Polymorphism and Aerobic Training on the Immune–Endocrine Network and Cardiovascular Parameters of Postmenopausal Women. J. Clin. Endocrinol. Metab. 2013, 98, 4187–4194. [Google Scholar] [CrossRef] [PubMed]
- Manzoli, L.; La Vecchia, C.; Flacco, M.E.; Capasso, L.; Simonetti, V.; Boccia, S.; Di Baldassarre, A.; Villari, P.; Mezzetti, A.; Cicolini, G. Multicentric cohort study on the long-term efficacy and safety of electronic cigarettes: Study design and methodology. BMC Public Health 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, M.; Izzicupo, P.; Santarelli, F.; Falone, S.; Pennelli, A.; Amicarelli, F.; Calafiore, A.M.; Di Baldassarre, A.; Gallina, S. ACE and AGTR1 Polymorphisms and Left Ventricular Hypertrophy in Endurance Athletes. Med. Sci. Sports Exerc. 2010, 42, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-S.V.; Kim, C.; Mercola, M. Electrophysiological challenges of cell-based myocardial repair. Circulation 2009, 120, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Lieu, D.K.; Liu, J.; Siu, C.-W.; McNerney, G.P.; Tse, H.-F.; Abu-Khalil, A.; Huser, T.; Li, R.A. Absence of transverse tubules contributes to non-uniform Ca2+ wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes. Stem Cells Dev. 2009, 18, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Binah, O.; Dolnikov, K.; Sadan, O.; Shilkrut, M.; Zeevi-Levin, N.; Amit, M.; Danon, A.; Itskovitz-Eldor, J. Functional and developmental properties of human embryonic stem cells-derived cardiomyocytes. J. Electrocardiol. 2007, 40, S192–S196. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-D.; Li, J.; Tweedie, D.; Yu, H.-M.; Chen, L.; Wang, R.; Riordon, D.R.; Brugh, S.A.; Wang, S.-Q.; Boheler, K.R.; et al. Crucial role of the sarcoplasmic reticulum in the developmental regulation of Ca2+ transients and contraction in cardiomyocytes derived from embryonic stem cells. FASEB J. 2006, 20, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Schiller, J.; Beyar, R.; Satin, J.; Gepstein, L. Calcium handling in embryonic stem cell-derived cardiac myocytes: Of mice and men. Ann. N. Y. Acad. Sci. 2006, 1080, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, T.G.; Minami, I.; Kurose, Y.; Yamauchi, K.; Tada, M.; Nakatsuji, N. Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem Cell Res. 2010, 4, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef] [PubMed]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.A.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Lahti, A.L.; Kujala, V.J.; Chapman, H.; Koivisto, A.-P.; Pekkanen-Mattila, M.; Kerkelä, E.; Hyttinen, J.; Kontula, K.; Swan, H.; Conklin, B.R.; et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis. Model. Mech. 2012, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.-Z.; Xie, Y.; Moyes, K.W.; Gold, J.D.; Askari, B.; Laflamme, M.A. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ. Res. 2010, 107, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.-H.; Butterfield, C.; Lin, R.-Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Kenyagin-Karsenti, D.; Snir, M.; Segev, H.; Amit, M.; Gepstein, A.; Livne, E.; Binah, O.; Itskovitz-Eldor, J.; Gepstein, L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Investig. 2001, 108, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Brito-Martins, M.; Harding, S.E.; Ali, N.N. beta(1)- and beta(2)-adrenoceptor responses in cardiomyocytes derived from human embryonic stem cells: Comparison with failing and non-failing adult human heart. Br. J. Pharmacol. 2008, 153, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Pillekamp, F.; Haustein, M.; Khalil, M.; Emmelheinz, M.; Nazzal, R.; Adelmann, R.; Nguemo, F.; Rubenchyk, O.; Pfannkuche, K.; Matzkies, M.; et al. Contractile properties of early human embryonic stem cell-derived cardiomyocytes: Beta-adrenergic stimulation induces positive chronotropy and lusitropy but not inotropy. Stem Cells Dev. 2012, 21, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, S.M.; Pointon, A.; Williams, A.W.; Cross, M.J.; Sidaway, J.E. Cardiac Non-myocyte Cells Show Enhanced Pharmacological Function Suggestive of Contractile Maturity in Stem Cell Derived Cardiomyocyte Microtissues. Toxicol. Sci. 2016, 152, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Han, Z.; Han, Z.C.; Li, Z. Proangiogenic Features of Mesenchymal Stem Cells and Their Therapeutic Applications. Stem Cells Int. 2016, 2016, 1314709. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; He, H.; Noiseux, N.; Liang, O.D.; Zhang, L.; Morello, F.; Mu, H.; Melo, L.G.; Pratt, R.E.; Ingwall, J.S.; et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006, 20, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Singla, D.K. Transplanted induced pluripotent stem cells mitigate oxidative stress and improve cardiac function through the Akt cell survival pathway in diabetic cardiomyopathy. Mol. Pharm. 2013, 10, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Merino, H.; Singla, D.K. Notch-1 mediated cardiac protection following embryonic and induced pluripotent stem cell transplantation in doxorubicin-induced heart failure. PLoS ONE 2014, 9, e101024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, X.; Liao, S.; Wang, W.; Wang, J.; Li, X.; Ding, Y.; Liang, Y.; Gao, F.; Yang, M.; et al. Potent Paracrine Effects of human induced Pluripotent Stem Cell-derived Mesenchymal Stem Cells Attenuate Doxorubicin-induced Cardiomyopathy. Sci. Rep. 2015, 5, 11235. [Google Scholar] [CrossRef] [PubMed]
- Bobis-Wozowicz, S.; Kmiotek, K.; Sekula, M.; Kedracka-Krok, S.; Kamycka, E.; Adamiak, M.; Jankowska, U.; Madetko-Talowska, A.; Sarna, M.; Bik-Multanowski, M.; et al. Human Induced Pluripotent Stem Cell-Derived Microvesicles Transmit RNAs and Proteins to Recipient Mature Heart Cells Modulating Cell Fate and Behavior. Stem Cells 2015, 33, 2748–2761. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-H.; Fu, X.; Yang, P.C. Exosomes Generated From iPSC-Derivatives: New Direction for Stem Cell Therapy in Human Heart Diseases. Circ. Res. 2017, 120, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, A.; Santoso, M.R.; Mahmoudi, M.; Shukla, P.; Wang, L.; Bennett, M.; Goldstone, A.B.; Wang, M.; Fukushi, M.; Ebert, A.D.; et al. Paracrine Effects of the Pluripotent Stem Cell-Derived Cardiac Myocytes Salvage the Injured Myocardium. Circ. Res. 2017, 121, e22–e36. [Google Scholar] [CrossRef] [PubMed]
- Powell, K. Stem-cell niches: It’s the ecology, stupid! Nature 2005, 435, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.L.; Moon, R.T.; Vunjak-Novakovic, G. It takes a village to grow a tissue. Nat. Biotechnol. 2005, 23, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Figallo, E.; Cannizzaro, C.; Elvassore, N.; Vunjak-Novakovic, G. Micro-bioreactor arrays for controlling cellular environments: Design principles for human embryonic stem cell applications. Methods 2009, 47, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Vunjak-Novakovic, G. Microscale technologies for regulating human stem cell differentiation. Exp. Biol. Med. 2014, 239, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Sirabella, D.; Yeager, K.; Davidson, K.; Simon, J.; Moon, R.T.; Vunjak-Novakovic, G. Microfluidic bioreactor for dynamic regulation of early mesodermal commitment in human pluripotent stem cells. Lab Chip 2013, 13, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Mandenius, C.-F.; Steel, D.; Noor, F.; Meyer, T.; Heinzle, E.; Asp, J.; Arain, S.; Kraushaar, U.; Bremer, S.; Class, R.; et al. Cardiotoxicity testing using pluripotent stem cell-derived human cardiomyocytes and state-of-the-art bioanalytics: A review: Cardiotoxicity testing using derived stem cells. J. Appl. Toxicol. 2011, 31, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.; Lai, B.F.L.; Lu, R.X.Z.; Korolj, A.; Wang, E.Y.; Radisic, M. High-Content Assessment of Cardiac Function Using Heart-on-a-Chip Devices as Drug Screening Model. Stem Cell Rev. 2017, 13, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, I.; Sun, B.; Lallos, G.; Terrenoire, C.; Freytes, D.O. Contributions of bone morphogenetic proteins in cardiac repair cells in three-dimensional in vitro models and angiogenesis. J. Tissue Eng. Regen. Med. 2018, 12, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Feric, N.T.; Radisic, M. Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv. Drug Deliv. Rev. 2016, 96, 110–134. [Google Scholar] [CrossRef] [PubMed]
- Eng, G.; Lee, B.W.; Protas, L.; Gagliardi, M.; Brown, K.; Kass, R.S.; Keller, G.; Robinson, R.B.; Vunjak-Novakovic, G. Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nat. Commun. 2016, 7, 10312. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5, 8883. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Vunjak-Novakovic, G.; Bhatia, S.; Chen, C.; Hirschi, K. HeLiVa platform: Integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther. 2013, 4 (Suppl. S1), S8. [Google Scholar] [CrossRef] [PubMed]
- Keung, W.; Ren, L.; Li, S.; Wong, A.O.-T.; Chopra, A.; Kong, C.-W.; Tomaselli, G.F.; Chen, C.S.; Li, R.A. Non-cell autonomous cues for enhanced functionality of human embryonic stem cell-derived cardiomyocytes via maturation of sarcolemmal and mitochondrial KATP channels. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Bellin, M.; Mummery, C.L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come?: Implementation of hiPSC-CMs in cardiotoxicity. Br. J. Pharmacol. 2017, 174, 3749–3765. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Kamalakar, A.; Yang, X.; Word, B.; Stockbridge, N.; Lyn-Cook, B.; Pang, L. Evaluation of Batch Variations in Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes from 2 Major Suppliers. Toxicol. Sci. 2017, 156, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, S.; Guo, L.; Mussio, J.; Furniss, M.; Hamre, J.; Davis, M. Examining the Protective Role of ErbB2 Modulation in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2014, 141, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.R.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharmacol. 2015, 285, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef] [PubMed]
- Pointon, A.; Harmer, A.R.; Dale, I.L.; Abi-Gerges, N.; Bowes, J.; Pollard, C.; Garside, H. Assessment of Cardiomyocyte Contraction in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2015, 144, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Lamore, S.D.; Ahlberg, E.; Boyer, S.; Lamb, M.L.; Hortigon-Vinagre, M.P.; Rodriguez, V.; Smith, G.L.; Sagemark, J.; Carlsson, L.; Bates, S.M.; et al. Deconvoluting Kinase Inhibitor Induced Cardiotoxicity. Toxicol. Sci. 2017, 158, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—Update on progress. J. Pharmacol. Toxicol. Methods 2016, 81, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Papoian, T. Moving beyond the comprehensive in vitro proarrhythmia assay: Use of human-induced pluripotent stem cell-derived cardiomyocytes to assess contractile effects associated with drug-induced structural cardiotoxicity: IPSC-CMs to assess contractile and structural cardiotoxicity. J. Appl. Toxicol. 2018. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, L.; Zeng, H.; White, S.L.; Furniss, M.; Balasubramanian, B.; Lis, E.; Lagrutta, A.; Sannajust, F.; Zhao, L.L.; et al. Multi-parametric assessment of cardiomyocyte excitation-contraction coupling using impedance and field potential recording: A tool for cardiac safety assessment. J. Pharmacol. Toxicol. Methods 2016, 81, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Honda, Y.; Watanabe, H.; Saiki, S.; Koyabu, K.; Itoh, T.; Nagasawa, C.; Nakamori, C.; Nakayama, C.; Iwasaki, H.; et al. CSAHi study-2: Validation of multi-electrode array systems (MEA60/2100) for prediction of drug-induced proarrhythmia using human iPS cell-derived cardiomyocytes: Assessment of reference compounds and comparison with non-clinical studies and clinical information. Regul. Toxicol. Pharmacol. 2017, 88, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Germanguz, I.; Sedan, O.; Zeevi-Levin, N.; Shtrichman, R.; Barak, E.; Ziskind, A.; Eliyahu, S.; Meiry, G.; Amit, M.; Itskovitz-Eldor, J.; et al. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell. Mol. Med. 2011, 15, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, U.; Nemade, H.; Gaspar, J.A.; Hescheler, J.; Hengstler, J.G.; Sachinidis, A. MicroRNAs as early toxicity signatures of doxorubicin in human-induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2016, 90, 3087–3098. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Node, K.; Fukuda, K. Cardiovascular Disease Modeling Using Patient-Specific Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2015, 16, 18894–18922. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Dell’Era, P. Cardiac disease modeling using induced pluripotent stem cell-derived human cardiomyocytes. World J. Stem Cells 2015, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.-S.; Schaniel, C.; Lee, D.-F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Martinez-Fernandez, A.; Yamada, S.; Perez-Terzic, C.; Ikeda, Y.; Terzic, A. Repair of Acute Myocardial Infarction by Human Stemness Factors Induced Pluripotent Stem Cells. Circulation 2009, 120, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Rojas, S.V.; Kensah, G.; Rotaermel, A.; Baraki, H.; Kutschka, I.; Zweigerdt, R.; Martin, U.; Haverich, A.; Gruh, I.; Martens, A. Transplantation of purified iPSC-derived cardiomyocytes in myocardial infarction. PLoS ONE 2017, 12, e0173222. [Google Scholar] [CrossRef] [PubMed]
- Place, E.S.; Evans, N.D.; Stevens, M.M. Complexity in biomaterials for tissue engineering. Nat. Mater. 2009, 8, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Cacciapuoti, I.; Parouchev, A.; Benhamouda, N.; Tachdjian, G.; Tosca, L.; et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical case report: Figure 1. Eur. Heart J. 2015, 36, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell–Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, P.J.; Horton, C.; Lahiri, P.; Shanmugarajah, K.; Davies, T.J. Beneath the sword of Damocles: Regenerative medicine and the shadow of immunogenicity. Regen. Med. 2016, 11, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Imberti, B.; Monti, M.; Casiraghi, F. Pluripotent stem cells and tolerance induction in organ transplantation. Curr. Opin. Organ Transplant. 2015, 20, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Kamao, H.; Iwasaki, Y.; Okamoto, S.; Hashiguchi, T.; Iseki, K.; Hayashi, N.; Mandai, M.; Takahashi, M. Inhibition of T-Cell Activation by Retinal Pigment Epithelial Cells Derived from Induced Pluripotent Stem Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Ghinassi, B.; Ferro, L.; Masiello, F.; Tirelli, V.; Sanchez, M.; Migliaccio, G.; Whitsett, C.; Kachala, S.; Riviere, I.; Sadelain, M.; et al. Recovery and Biodistribution of Ex Vivo Expanded Human Erythroblasts Injected into NOD/SCID/IL2R γ null mice. Stem Cells Int. 2011, 2011, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qin, J.; Zhao, R.C.; Zenke, M. Reduced Immunogenicity of Induced Pluripotent Stem Cells Derived from Sertoli Cells. PLoS ONE 2014, 9, e106110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, P.; Meyer, E.H.; Kooreman, N.G.; Diecke, S.; Dey, D.; Sanchez-Freire, V.; Hu, S.; Ebert, A.; Odegaard, J.; Mordwinkin, N.M.; et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.; Hyllner, J.; Stacey, G.; Taylor, C.J.; Turner, M. Setting up a Haplobank: Issues and Solutions. Curr. Stem Cell Rep. 2015, 1, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Burridge, P.W.; Yu, K.-H.; Ahrens, J.H.; Termglinchan, V.; Wu, H.; Liu, C.; Shukla, P.; Sayed, N.; Churko, J.M.; et al. Transcriptome Profiling of Patient-Specific Human iPSC-Cardiomyocytes Predicts Individual Drug Safety and Efficacy Responses In Vitro. Cell Stem Cell 2016, 19, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Epigenetic Modifications | Name | Action | Reference |
|---|---|---|---|
| Histone acetylation | Histone acetyltransferase (HAT) | P300 is essential for cardiac development. It contributes to Gata4, Srf, Mef5c expression. P300 knockout mice are embryonically lethal | [37] |
| Histone deacetylase (HDAC) | Mice lacking both HDAC1 and HDAC2 show neonatal lethality due to arrhythmias and dilated cardiomyopathy | [38] | |
| Inhibitors of HDAC | Trichostatin A promotes cardiac differentiation increasing expression of Gata4, Mef2c and Nkx2.5 | [25] | |
| H3K9ac and H3K27ac | In CMs from fetal to adult stages, TNNT2 shows a sequential enrichment of active histone markers such as H3K9ac and H3K27ac | [24] | |
| Histone methylation | Histone methyltransferases (HTMs) | Loss of HMT Smyd1 is embryonic lethal, because mice show right ventricular hypoplasia and impaired cardiomyocyte maturation. | [21] |
| HTM WHSC1 is involved in Nkx2.5 repression via H3K3me37. | |||
| Histone demethylase (HDMs) | The HDM UTX removes H3K29me3 activating the cardiac transcription factors Gata4, Nkx2.5, Srf, Tbx5. Mice lacking UTX show severe heart malformation. | [20] | |
| H3K4me and H3K27 | H3K4 methylation levels are fundamental in murine CMs. A loss of H3K4 methylation can result in intracellular calcium modifications and increased contractility | [18] | |
| FGF19 and NODAL genes show high levels of H3K4me3 and H3K27me3 in undifferentiated ESC and low levels during the differentiation | [17] | ||
| Cardiac transcription factorsGata4, Wnt2, Tbx2, Nkx2.5 show high levels H3K27me3 during the pluripotency that decrease during differentiation, in the same time there is a gradual increase in H3K36me3 and H3K4me3 | |||
| Wnt, Hedgehog, TGFβ family, VEGF, FGF family, PDGF(pathways involved in cardiac differentiation) show a stage-specific repression by H3K27me3 and activation by H3K36me3 and H3K4me3 | |||
| DNA methylation | DNA methyl transferase (DNM) | DNMT1 expression decreases from mesoderm to CM stage while DNMT3A increases from ESC to primitive mesoderm stage. WNT and TGF-β genes undergo promoter methylation changes, the latter pathway became hypomethylated and upregulated in CM stage, whereas generally WNT genes acquire promoter methylation | [22] |
| Inhibitors of DNA methylation | 5-Azacytidine promotes cardiac differentiation in ES and adult mesenchymal stem cells | [25] | |
| mCpG | In CMs from fetal to adult stages, TNNT2 shows a sequential loss of mCpG, instead fetally expressed TNNI1 is silenced postnatally and there is a loss of de novo mCPG. | [24] | |
| Comparison of mCpG changes during development of fetal and maturation of infantile CMs showed a predominant loss of mCpG | |||
| Changes in mCpG is accompanied by changes in histone marks. Demethylated region during maturation gained the active histone marks H3K27ac, H3K4me3, H3K36me3 and H3K9ac, whereas hypermethylated region showed a loss of these marks | |||
| Long non-coding RNA | Braveheart | Braveheart is an activator of Mesp1, Gata4, Nkx2,5, TBx5, Hand1. Braveheart acts upstream Mesp1 and regulates the temporal activation of cardiac genes through modulation of Mesp1 itself | [27] |
| Braveheart interacts with SUZ12 that acts as a histone methyltransferase. | |||
| Braveheart induces the differentiation of murine bone-marrow-derived mesenchymal cells into cells with a cardiogenic phenotype.It increases sarcomeric α-actin and cardiac troponin T expression and the upregulation of Gata4, Nkx2.5, Isl-1 and Mesp1. | [28] | ||
| Fendrr | Fendrr Interacts with PRC2 and Trg/MLL complex to modulate the chromatin signature of pitX2 and Foxf1. | [29] | |
| Loss of Fendrr affects the expression of Nkx2.5 and Gata4. Fendrr knockout is embryonic lethal in mice due to defect on the heart septum. | [29] | ||
| MicroRNAs | miR-1, miR-499 | miR-1 controls myogenic differentiation in mouse heart | [30] |
| miR-499 is a cardiac specific miRNA | |||
| miR-1 and miR-499 enhance the cardiac differentiation of cardiomyocyte progenitor cells, probably targeting Sox6 with a consequent increasing of α-cardiac actinin and cardiac troponin T | |||
| Inhibition of miR-1 and miR-499 blocks cardiac differentiation. | |||
| miR-322/-503 cluster | miR-322/-503 cluster encodes in an intergenic region on the X-chromosome and increases Nkx2.5, Mef2c, Tbx5, α-MHC inducing CM differentiation, probably targeting Celf1, whereas their deletion reduces the expression of cardiac markers | [33] | |
| miR-322/-503 cluster acts by the repression of their target Celf1, that lead the ESC to the neuronal differentiation: it is likely that the miR-322/-503 cluster promotes the cardiac differentiation impairing the neuronal through Celf1 inhibition | |||
| miR-208 | miR-208 is involved in the regulation of myosin heavy chain isoform switch during developmental and pathophysiological condition. | [35] | |
| miR-1-2 | miR-1-2 induces cardiac differentiation of murine bone marrow-derived mesenchymal stem cells by Wnt signaling pathway | [34] | |
| Transfection with miR-1-2 increases expression of Nkx2.5, Gata4, cTnI | |||
| miR-133 | miR-133 together with Gata4, Tbx5 and Mef2c improves cardiac reprogramming from human or murine fibroblast, by repressing Snai1 | [30,32] | |
| miR-26b | miR-26b promotes cardiac differentiation of P19 cells, by regulating canonical and non-canonical Wnt pathway. It represses the expression of Wnt5a and Gsk3β | [31] | |
| let-7 | let-7 family is upregulated during in vitro human cardiac differentiation. | [36] | |
| The overexpression of members of let-7 family for 2 weeks in hESC derived CMs increases contractile force, cell size, sarcomere length and action potential duration. Knockdown of let-7 results in a reduction of sarcomere length and expression of cardiac maturation markers. Let-7 family probably acts downregulation two of its targets, IRS2 (a member of insulin signaling pathway) and EZH2 (a histone methyltransferase that can regulate gene expression) |
| Differentiation Condition | Inductive Factors | Beating Starting | Efficiency | CM Subtypes | Functional Assays | Ref |
|---|---|---|---|---|---|---|
| EB Formation-Based Culture | ||||||
| Static suspension culture | Activin A, BMP4, VEGF, DKK1, bFGF, Ascorbic Acid | Day 10 | 40–50% (cTNT day 14–16) | atrial, ventricular | Extracellular electrical activity, Patch clamp analysis, Cell transplantation | [45] |
| Static suspension culture | Activin A, BMP4, IWR-1, Ascorbic Acid, Blebbistatin | Day 7 | 100% beating EBs day 15 90% cTNT day 21 | Ventricular | Extracellular electrical activity, Patch clamp analysis, Optical mapping of membrane potential | [46] |
| Forced aggregation (96 well) | Activin A, BMP4, VEGF, SCF, WNT3a | Day 9 | 96% beating EBs day 10 27% Nkx2.5 day 10 | n.a. | Extracellular electrical activity, Patch clamp analysis | [47] |
| Forced aggregation (96 well and AggreWell) | Activin A, BMP4, bFGF, Lipids, Insulin, CHIR, IWP-2 | Day 6 | 100% beating EBs day 6 50% cTnT day 6 | ventricular | Extracellular electrical activity, Patch clamp analysis, Intracellular calcium transient imaging | [48] |
| Monolayer Culture | ||||||
| Monolayer | Activin A, BMP4 | Day 12 | 50% MHC day 21 | n.a. | Transplantation to the heart | [49] |
| Monolayer-sandwich | Activin A, BMP4, bFGF | Day 7 | 90% cTnT day 30 | Mixed | Patch clamp analysis, Intracellular calcium transient imaging | [50] |
| Monolayer | BMP4, bFGF, CHIR, IWP-2, Ascorbic Acid | Day 6 | 90% cTnT | ventricular | Extracellular electrical activity, Patch clamp analysis, Intracellular calcium transient imaging | [48] |
| Monolayer | CHIR, IWP-2 | Day 7 | 98% cTnT day 15 | Mixed (atrial and ventricular) | Patch clamp analysis | [51] |
| Monolayer | CHIR, WNT-C59, Ascorbic Acid | Day 7 | 90% cTnT | Mixed | Extracellular electrical activity-based nanopillar recording, Patch clamp analysis | [52] |
| Monolayer | CHIR99021, IWR-1, T3, Dexamethasone | 80% colocalization of sarcomeric alpha actinin and Junctophilin 2 | Mixed | T Tubule staining, Paced Calcium Transients, Calcium Kinetics and contractility | [53] | |
| Suspension Large Scale Culture | ||||||
| Matrix-dependent aggregates/Rocker culture | CHIR, IWP2 | Day 7 | 65% (cTnT/day 12) | n.a. | Toxicology assay | [54] |
| Matrix-dependent aggregates/EB formation/spinner flasks | SB203580 | Day 10 | 80% (beating EBs/day 16) 20% (MHC/day 16) | n.a. | QT prolongation assay and CM toxicity test | [55] |
| Matrix-independent aggregated/Erlenmeyer Flask and bioreactor | CHIR, IWP2 | Day 6–7 | 84% (cTnT, MHC/day 10) | 80–90% ventricular | Bioartificial cardiac tissue generation, Patch clamp analysis, Extracellular electrical activity | [56] |
| Matrix-independent aggregated/Spinner flask | CHIR, IWR-1, SB431542, Purmorphamine | Day 7 | >90% (cTnT/day 15) | n.a. | Extracellular electrical activity, Patch clamp analysis | [57] |
| Adult-CM | hPSC-CM | ||
|---|---|---|---|
| Beating | Quiescent | Present | |
| Conduction Properties | Capacitance | 150 pF | 20–50 pF |
| Resting mem potential | −80 to −90 mV | −20 to −60 mV | |
| Upstroke velocity | 150–350 V/s | 10–50 V/s | |
| Conduction velocity | 60 cm/s | 10–20 cm/s | |
| Location of gap junctions | Intercalated discs | Circumference of cells | |
| Ion channel density (pA/pF) | INa | −196 | −100 to −244 |
| ICaL | −4.3 to −10.2 | −2.2 to −10 | |
| Ito | 2.3 to 10.6 | 2.5 to 13.7 | |
| IKs | 0.18 to 0.58 | 0.3 to 0.7 | |
| IKr | 0.5 | 0.4 to 0.8 | |
| IK1 | −12 | 0 to −3.4 | |
| INCX | 2.5 to 3 | 3.6 to 7.9 (inward mode) | |
| Ca2+ kinetics | APD90 | 260 ms | 300–700 ms |
| Cycle Length | 0.8–1 s | 0.8–2 s | |
| T-rise | 2.5 ms | 3.5–10 ms | |
| Triangulation | 45 ms | 45–120 ms | |
| Drug | Cardiac Safety Index |
|---|---|
| Afatinib | 0.444 |
| Erlotinib | 0.635 |
| Gefitinib | 0.409 |
| Lapatinib | 0.209 |
| Axitinib | 1.000 |
| Cabozatinib | 0.769 |
| Pazopanib | 0.671 |
| Ponatinib | 0.483 |
| Regorafenib | 0.010 |
| Sorafenib | 0.004 |
| Sunitinib | 0.218 |
| Vandetanib | 0.041 |
| Bosutinib | 0.315 |
| Dasatinib | 0.524 |
| Imatinib | 0.126 |
| Nilotinib | 0.104 |
| Dabrafenib | 0.459 |
| Vemurafenib | 0.003 |
| Trametinib | 1.000 |
| Ibrutinib | 0.507 |
| Crizotinib | 0.063 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Baldassarre, A.; Cimetta, E.; Bollini, S.; Gaggi, G.; Ghinassi, B. Human-Induced Pluripotent Stem Cell Technology and Cardiomyocyte Generation: Progress and Clinical Applications. Cells 2018, 7, 48. https://doi.org/10.3390/cells7060048
Di Baldassarre A, Cimetta E, Bollini S, Gaggi G, Ghinassi B. Human-Induced Pluripotent Stem Cell Technology and Cardiomyocyte Generation: Progress and Clinical Applications. Cells. 2018; 7(6):48. https://doi.org/10.3390/cells7060048
Chicago/Turabian StyleDi Baldassarre, Angela, Elisa Cimetta, Sveva Bollini, Giulia Gaggi, and Barbara Ghinassi. 2018. "Human-Induced Pluripotent Stem Cell Technology and Cardiomyocyte Generation: Progress and Clinical Applications" Cells 7, no. 6: 48. https://doi.org/10.3390/cells7060048
APA StyleDi Baldassarre, A., Cimetta, E., Bollini, S., Gaggi, G., & Ghinassi, B. (2018). Human-Induced Pluripotent Stem Cell Technology and Cardiomyocyte Generation: Progress and Clinical Applications. Cells, 7(6), 48. https://doi.org/10.3390/cells7060048