Characterization of Carotid Smooth Muscle Cells during Phenotypic Transition


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Human Vascular Smooth Muscle Cells
2.2. Flow Cytometry
2.3. Gene Expression Analysis
2.4. Western Blot
2.5. Confocal Microscopy
3. Results
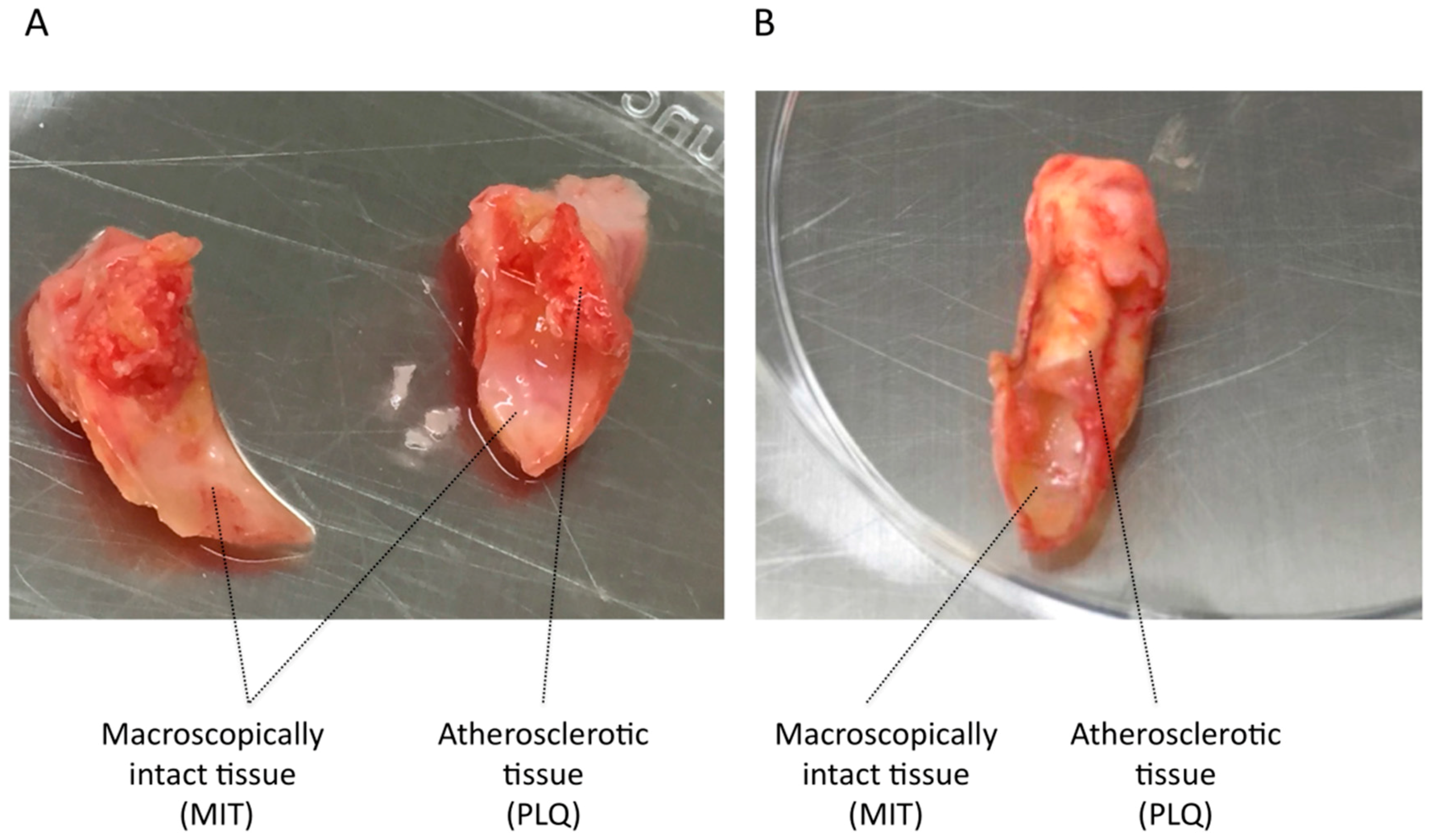
3.1. VSMC Isolation from Human Carotid Atheroma Plaque
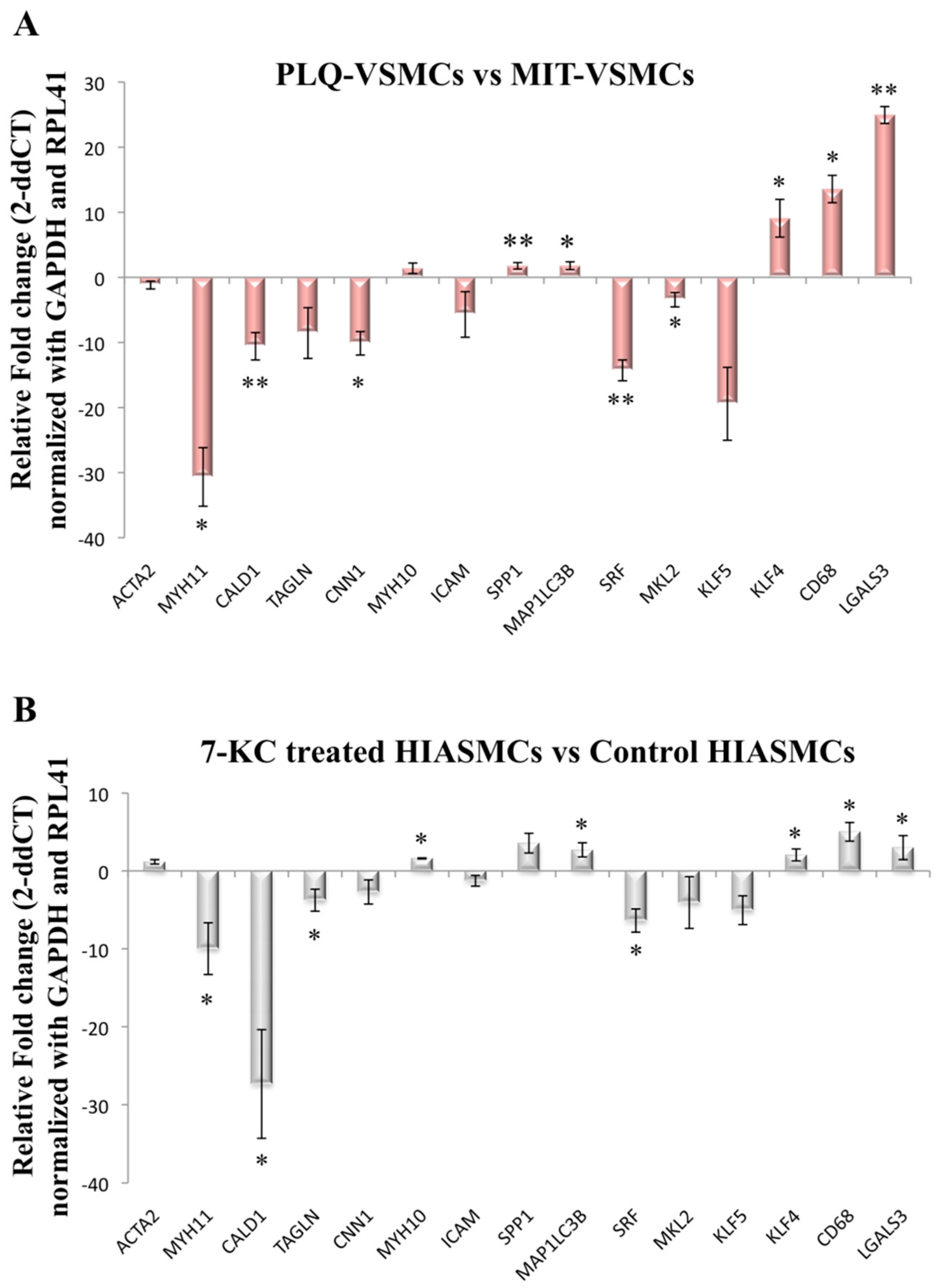
3.2. VSMC-Specific Gene Expression Pattern Shows Phenotypic Modulation in VSMCs Isolated from Human Carotid Plaques
3.3. MYH1, SPP1, and KLF5 Emerge as Differentially Expressed Genes between Symptomatic and Asymptomatic Carotid VSMC
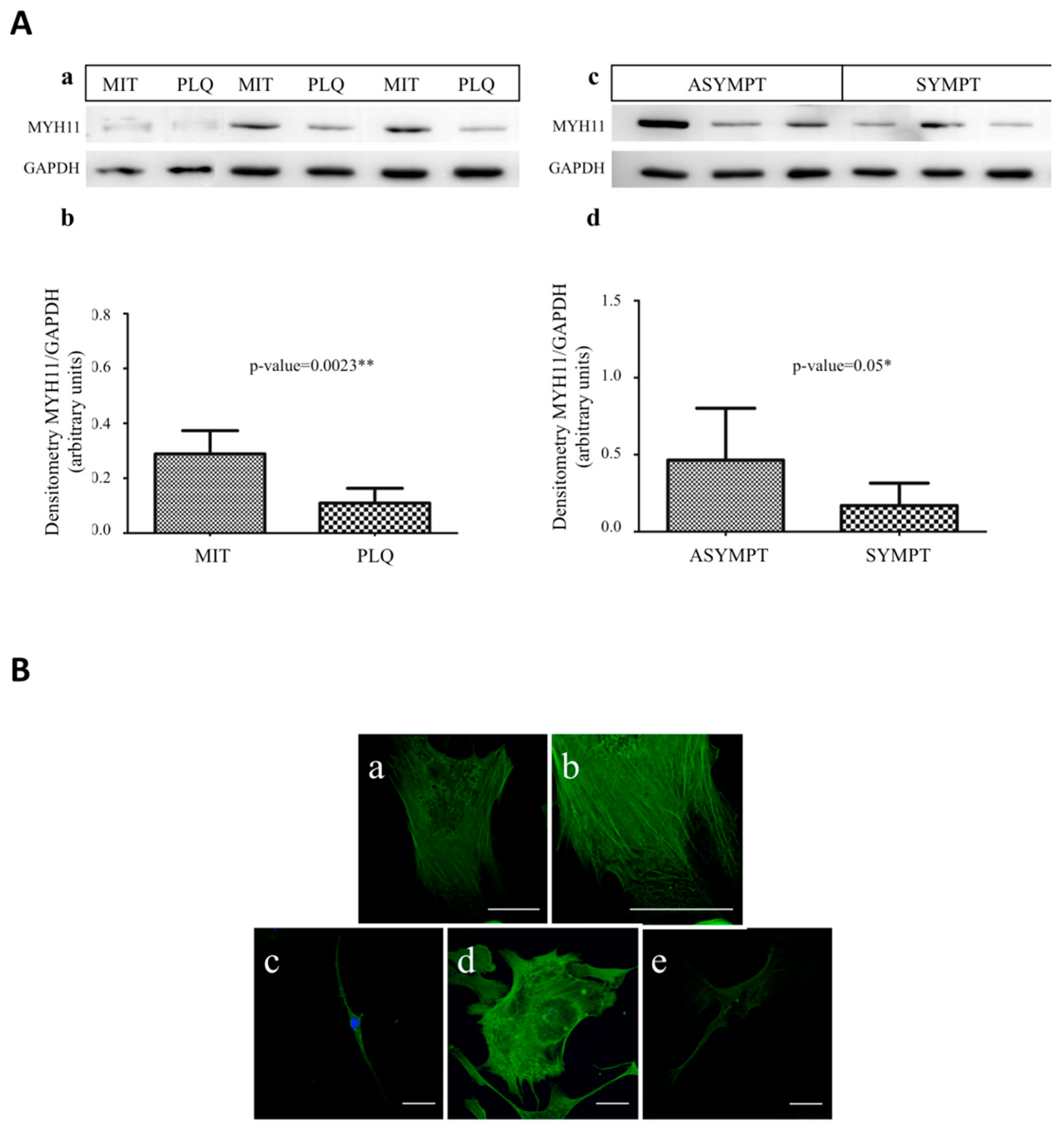
3.4. MYH11 Expression in VSMCs from Symptomatic vs. Asymptomatic Patients and from PLQ vs. MIT Region
3.5. MMP3, MMP7, MMP9, and TIMP1 Do Not Emerge as Differentially Expressed in VSMCs Coming from Asymptomatic and Symptomatic Patients or between PLQ and MIT Area
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carr, S.; Farb, A.; Pearce, W.H.; Virmani, R.; Yao, J.S.T. Atherosclerotic plaque rupture in symptomatic carotid artery stenosis. J. Vasc. Surg. 1996, 23, 755–766. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic Plaque Destabilization. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Greenhalgh, R.M.; Davies, A.H. The Symptomatic Carotid Plaque. Stroke 2000, 31, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Colio, L.M.; Martín-Ventura, J.L.; Vivanco, F.; Michel, J.-B.; Meilhac, O.; Egido, J. Biology of atherosclerotic plaques: what we are learning from proteomic analysis. Cardiovasc. Res. 2006, 72, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, C.; Coen, M.; Bochaton-Piallat, M.-L. Smooth muscle cell phenotypic switch: implications for foam cell formation. Curr. Opin. Lipidol. 2014, 25, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Virmani, R.; Rosenfeld, M.E. The good smooth muscle cells in atherosclerosis. Curr. Atheroscler. Rep. 2000, 2, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth Muscle Cell Plasticity: Fact or Fiction? Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, Y.Q.; Wang, X.; Pi, Y.; Gao, C.-Y.; Li, J.-C.; Zhang, L.-L. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem. Cell Biol. 2016, 145, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Alloza, I.; Goikuria, H.; Idro, J.L.; Triviño, J.C.; Fernández Velasco, J.M.; Elizagaray, E.; García-Barcina, M.; Montoya-Murillo, G.; Sarasola, E.; et al. RNAseq based transcriptomics study of SMCs from carotid atherosclerotic plaque: BMP2 and IDs proteins are crucial regulators of plaque stability. Sci. Rep. 2017, 7, 3470. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.-B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Rocchiccioli, S.; Ucciferri, N.; Comelli, L.; Trivella, M.G.; Citti, L.; Cecchettini, A. Proteomics changes in adhesion molecules: A driving force for vascular smooth muscle cell phenotypic switch. Mol. Biosyst. 2012, 8, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Kundu, G.C.; Lee, B.I.; Peri, A.; Fohl, D.; Chackalaparampil, I.; Mukherjee, B.B.; Li, X.D.; Mukherjee, D.C.; Seides, S.; et al. Potential roles of osteopontin and αVβ3 integrin in the development of coronary artery restenosis after angioplasty. Proc. Natl. Acad. Sci. USA 1997, 94, 9308–9313. [Google Scholar] [CrossRef] [PubMed]
- Worth, N.F.; Rolfe, B.E.; Song, J.; Campbell, G.R. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cytoskeleton 2001, 49, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Meena, S.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2014, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Miano, J.M.; Cserjesi, P.; Olson, E.N. SM22α, a Marker of Adult Smooth Muscle, Is Expressed in Multiple Myogenic Lineages during Embryogenesis. Circ. Res. 1996, 78, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Sigala, F.; Oikonomou, E.; Antonopoulos, A.S.; Galyfos, G.; Tousoulis, D. Coronary versus carotid artery plaques. Similarities and differences regarding biomarkers morphology and prognosis. Curr. Opin. Pharmacol. 2018, 39, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Schaar, J.A.; Muller, J.E.; Falk, E.; Virmani, R.; Fuster, V.; Serruys, P.W.; Colombo, A.; Stefanadis, C.; Ward Casscells, S.; Moreno, P.R.; et al. Terminology for High-Risk and Vulnerable Coronary Artery Plaques. Eur. Heart J. 2004, 25, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biology. 2002, 3. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis Novelty and Significance. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Worth, N.F.; Rolfe, B.E.; Campbell, G.R.; Campbell, J.H. Heterogeneous distribution of isoactins in cultured vascular smooth muscle cells does not reflect segregation of contractile and cytoskeletal domains. J. Histochem. Cytochem. 2000, 48, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Pipes, G.C.T.; Creemers, E.E.; Olson, E.N. The Myocardin Family of Transcriptional Coactivators: Versatile Regulators of Cell Growth, Migration, and Myogenesis. Genes Dev. 2006, 20, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. Atherosclerosis and the Arterial Smooth Muscle Cell. Science 1973, 180, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef] [PubMed]
- Arkenbout, E.K.; Dekker, R.J.; de Vries, C.J.M.; Horrevoets, A.J.G.; Pannekoek, H. Focusing on transcription factor families in atherogenesis: The function of LKLF and TR3. Thromb. Haemost. 2003, 89, 522–529. [Google Scholar] [PubMed]
- Zheng, B.; Han, M.; Wen, J.K. Role of Krüppel-like Factor 4 in Phenotypic Switching and Proliferation of Vascular Smooth Muscle Cells. IUBMB Life 2010, 62, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Manabe, I.; Fujiu, K.; Yamamoto, T.; Takeda, N.; Eguchi, K.; Furuya, A.; Kuro-o, M.; Sata, M.; Nagai, R. Bone marrow—Derived cells contribute to vascular inflammation but do not differentiate into smooth muscle cell lineages. Circulation 2010, 122, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, B.; Zhao, Y.; Xie, S.; Nie, R. Differentiated markers in undifferentiated cells: Expression of smooth muscle contractile proteins in multipotent bone marrow mesenchymal stem cells. Dev. Growth Differ. 2013, 55, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Rolfe, B.E.; Campbell, J.H.; Campbell, G.R. Changes in three-dimensional architecture of microfilaments in cultured vascular smooth muscle cells during phenotypic modulation. Tissue Cell 1998, 30, 324–333. [Google Scholar] [CrossRef]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Saltis, J.; Thomas, A.C.; Agrotis, A.; Campbell, J.H.; Campbell, G.R.; Bobik, A. Expression of growth factor receptors on arterial smooth muscle cells. Dependency on cell phenotype and serum factors. Atherosclerosis 1995, 118, 77–87. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Sandison, M.E.; Dempster, J.; McCarron, J.G. The transition of smooth muscle cells from a contractile to a migratory, phagocytic phenotype: direct demonstration of phenotypic modulation. J. Physiol. 2016, 594, 6189–6209. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; Hill, B.G. Autophagic regulation of smooth muscle cell biology. Redox Biol. 2015, 4, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; López Medina, A.; Del Mar Freijo, M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M. Cholesterol loading reprograms the miR-143/145–myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Huff, M.W.; Pickering, J.G. Can a Vascular Smooth Muscle–Derived Foam-Cell Really Change its Spots? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 492–495. [Google Scholar] [CrossRef] [PubMed][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Asymptomatic | Symptomatic | p-Value |
|---|---|---|---|
| n | 19 | 20 | |
| Age | 68 ± 9 | 71 ± 8 | ns (0.5) |
| Sex | 14 male/5 female | 16 male/4 female | ns (0.6) |
| Risk Factors (%) | |||
| Contralateral occlusion | 66 | 61 | ns (1.0) |
| Hypertension | 66 | 69 | ns (1.0) |
| Diabetes mellitus | 51 | 8 | ns (0.09) |
| Augmented cholesterol | 83 | 62 | ns (0.3) |
| Cardiopathy | 0 | 23 | ns (0.2) |
| Ischemic cardiopathy | 41 | 46 | ns (1.0) |
| Atrial fibrillation | 9 | 23 | ns (0.6) |
| Intermittent claudication | 26 | 24 | ns (1.0) |
| Tobacco | 16 | 8 | ns (0.6) |
| Medications (%) | |||
| Statins | 100 | 70 | ns (0.1) |
| Anticoagulant | 8 | 23 | ns (0.2) |
| Gene Symbol | FC (Asympt vs. Sympt) | p Value |
|---|---|---|
| Actin, alpha 2, smooth muscle, aorta (ACTA2) | 1.3 | ns |
| CD68 molecule (CD68) | −1.01 | ns |
| Caldesmon 1 (CALD1) | −1.17 | ns |
| Calponin 1 (CNN1) | 1.14 | ns |
| Galectin 3 (LGALS3) | 1.05 | ns |
| Intercellular adhesion molecule 1 (ICAM1) | −1.30 | ns |
| Kruppel like factor 4( KLF4) | 1.04 | ns |
| Kruppel like factor 5 (KLF5) | −1.89 | 0.01 |
| Microtubule associated protein 1 light chain 3 beta | −1.09 | ns |
| MKL1/myocardin like 2 (MKL2) | −1.17 | ns |
| Myosin heavy chain 10 (MYH10) | −1.31 | ns |
| Myosin heavy chain 11 (MYH11) | −4.53 | 0.045 |
| Secreted phosphoprotein 1 (SPP1) | 2.08 | 0.05 |
| Serum response factor (SRF) | −1.38 | ns |
| Transgelin (TAGLN) | −1.01 | ns |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goikuria, H.; Freijo, M.D.M.; Vega Manrique, R.; Sastre, M.; Elizagaray, E.; Lorenzo, A.; Vandenbroeck, K.; Alloza, I. Characterization of Carotid Smooth Muscle Cells during Phenotypic Transition. Cells 2018, 7, 23. https://doi.org/10.3390/cells7030023
Goikuria H, Freijo MDM, Vega Manrique R, Sastre M, Elizagaray E, Lorenzo A, Vandenbroeck K, Alloza I. Characterization of Carotid Smooth Muscle Cells during Phenotypic Transition. Cells. 2018; 7(3):23. https://doi.org/10.3390/cells7030023
Chicago/Turabian StyleGoikuria, Haize, Maria Del Mar Freijo, Reyes Vega Manrique, María Sastre, Elena Elizagaray, Ana Lorenzo, Koen Vandenbroeck, and Iraide Alloza. 2018. "Characterization of Carotid Smooth Muscle Cells during Phenotypic Transition" Cells 7, no. 3: 23. https://doi.org/10.3390/cells7030023
APA StyleGoikuria, H., Freijo, M. D. M., Vega Manrique, R., Sastre, M., Elizagaray, E., Lorenzo, A., Vandenbroeck, K., & Alloza, I. (2018). Characterization of Carotid Smooth Muscle Cells during Phenotypic Transition. Cells, 7(3), 23. https://doi.org/10.3390/cells7030023