Impaired Cargo Clearance in the Retinal Pigment Epithelium (RPE) Underlies Irreversible Blinding Diseases


Abstract
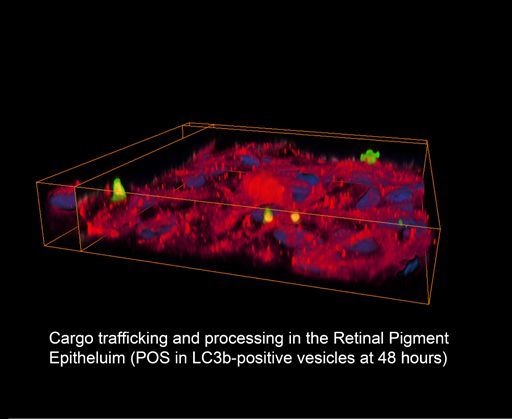
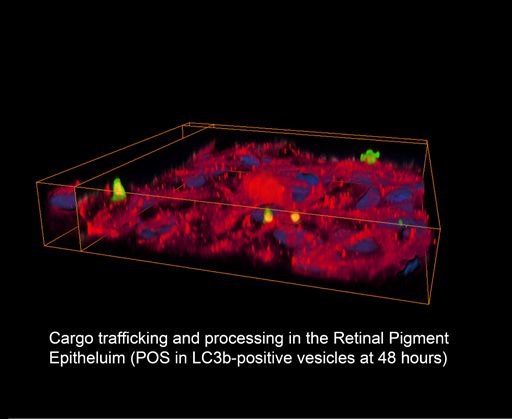{kind=link}
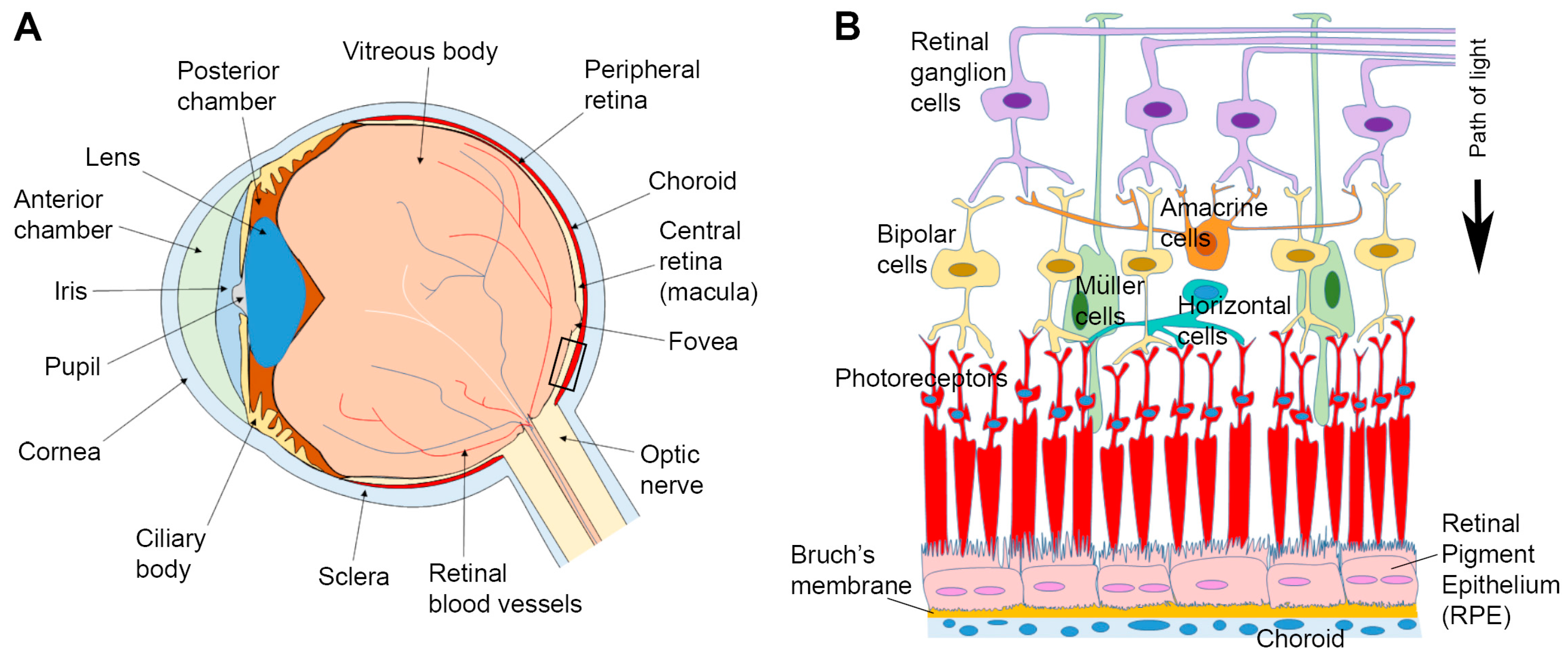{kind=link}
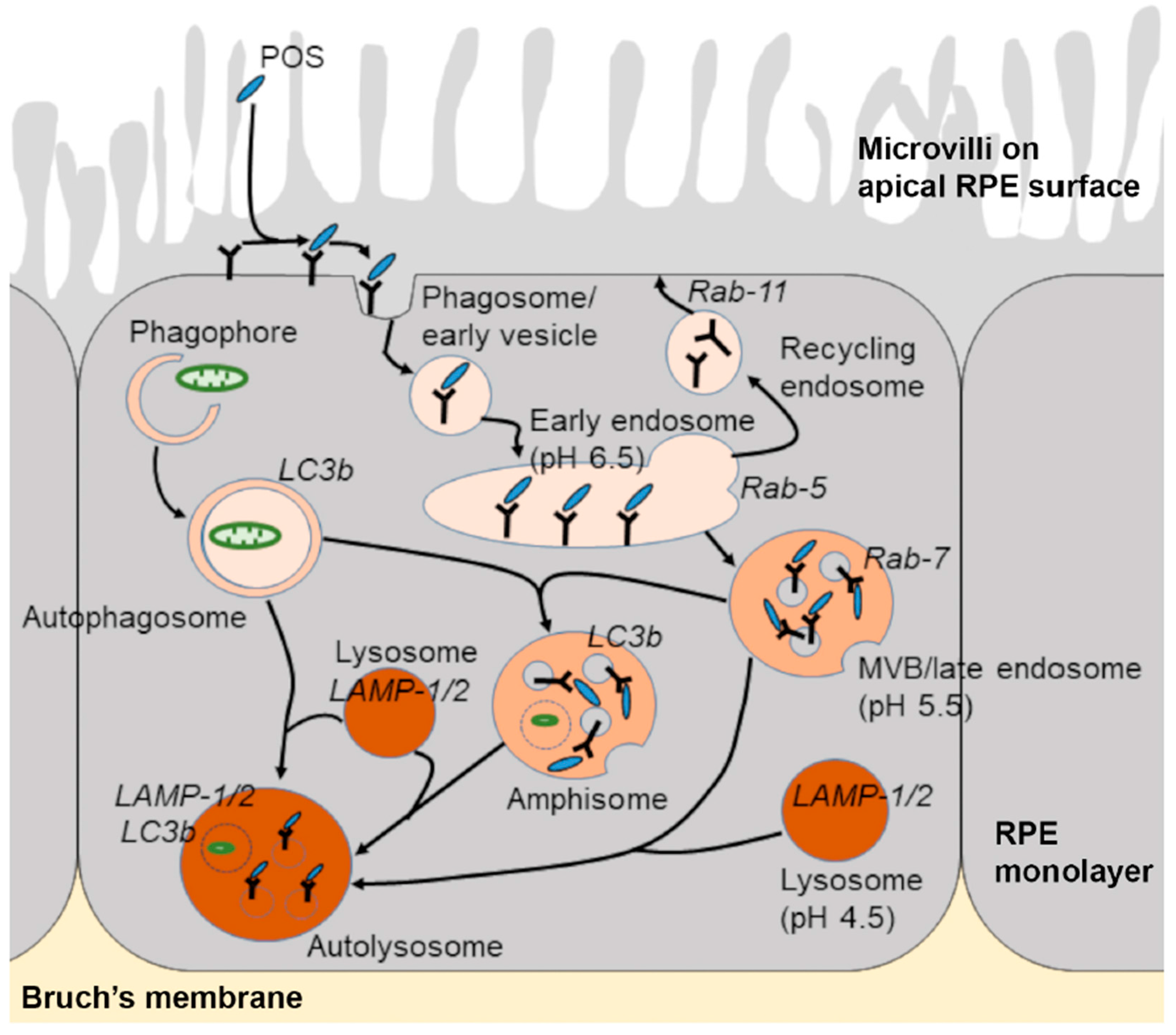{kind=link}
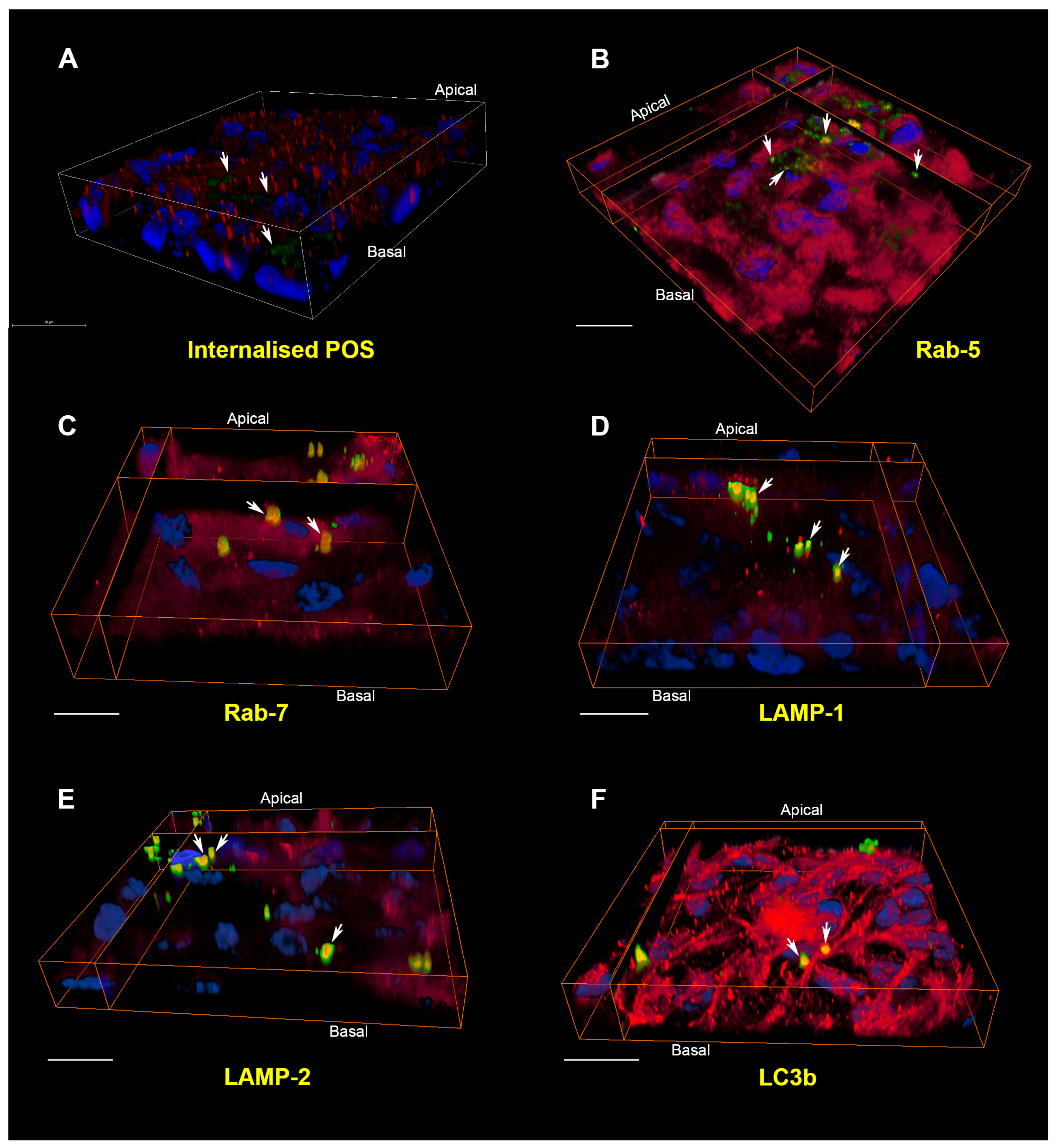{kind=link}
1. Introduction
2. Components of the Endocytic/Phagosome and Autophagy Pathways
2.1. The Endo-Lysosomal Pathway
2.2. The Phagocytic Pathway
2.3. The Autophagy Pathway
3. Impaired Cargo Handling and Proteolysis Underlies Several Chronic Degenerative Diseases
4. RPE Impairment and Retinal Disease
5. Impairment of Lysosomes and other Components of Cargo Handling in RPE Cells
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Young, R.W. The renewal of rod and cone outer segments in the rhesus monkey. J. Cell Biol. 1971, 49, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Bok, D. The retinal pigment epithelium: A versatile partner in vision. J. Cell Sci. 1993, 1993, 189–195. [Google Scholar] [CrossRef]
- Sparrrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Anderson, D.H.; Fisher, S.K. The relationship of primate foveal cones to the pigment epithelium. J. Ultrastruct. Res. 1979, 67, 23–32. [Google Scholar] [CrossRef]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Khandhadia, S.; Cherry, J.; Lotery, A.J. Age-related macular degeneration. In Neurodegenerative Diseases; Springer: New York, NY, USA, 2012; pp. 15–36. [Google Scholar]
- Ferrington, D.A.; Sinha, D.; Kaarniranta, K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 2016, 51, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PloS ONE 2009, 4, e4160. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, P.E.; Sarks, S.H.; Daw, N.; Constable, I.J. Distribution of cathepsin d in human eyes with or without age-related maculopathy. Exp. Eye Res. 1999, 69, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.G.; Ablonczy, Z.; Koutalos, Y.; Hanneken, A.M.; Spraggins, J.M.; Calcutt, M.W.; Crouch, R.K.; Caprioli, R.M.; Schey, K.L. Bis(monoacylglycero)phosphate lipids in the retinal pigment epithelium implicate lysosomal/endosomal dysfunction in a model of stargardt disease and human retinas. Sci. Rep. 2017, 7, 17352. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Yin, J.; Winborn, C.S.; Zhang, Q.; Yue, J.; Chaum, E. Prominin-1 is a novel regulator of autophagy in the human retinal pigment epithelium. Investig. Ophthalmol. Visual Sci. 2017, 58, 2366–2387. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.X.; Toops, K.A.; Lakkaraju, A. Protective responses to sublytic complement in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2016, 113, 8789–8794. [Google Scholar] [CrossRef] [PubMed]
- Wavre-Shapton, S.T.; Tolmachova, T.; Lopes da Silva, M.; Futter, C.E.; Seabra, M.C. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS ONE 2013, 8, e57769. [Google Scholar] [CrossRef]
- Roberts, R.L.; Barbieri, M.A.; Pryse, K.M.; Chua, M.; Morisaki, J.H.; Stahl, P.D. Endosome fusion in living cells overexpressing gfp-rab5. J. Cell Sci. 1999, 112, 3667–3675. [Google Scholar] [PubMed]
- Nielsen, E.; Severin, F.; Backer, J.M.; Hyman, A.A.; Zerial, M. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1999, 1, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Endocytosis and the recycling of plasma membrane. J. Cell Biol. 1983, 96, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Villarroel-Campos, D.; Gastaldi, L.; Conde, C.; Caceres, A.; Gonzalez-Billault, C. Rab-mediated trafficking role in neurite formation. J. Neurochem. 2014, 129, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harbor Perspect. Biol. 2014, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maxson, M.E.; Grinstein, S. The vacuolar-type h(+)-atpase at a glance—More than a proton pump. J. Cell Sci. 2014, 127, 4987–4993. [Google Scholar] [CrossRef] [PubMed]
- Sachse, M.; Urbe, S.; Oorschot, V.; Strous, G.J.; Klumperman, J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell 2002, 13, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The escrt machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Collinet, C.; Stoter, M.; Bradshaw, C.R.; Samusik, N.; Rink, J.C.; Kenski, D.; Habermann, B.; Buchholz, F.; Henschel, R.; Mueller, M.S.; et al. Systems survey of endocytosis by multiparametric image analysis. Nature 2010, 464, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. Embo. J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Yamashiro, D.J. Endosome acidification and the pathways of receptor-mediated endocytosis. Adv. Exp. Med. Biol. 1987, 225, 189–198. [Google Scholar] [PubMed]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin a1 and nocodazole on endocytic transport in hela cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [PubMed]
- Driskell, O.J.; Mironov, A.; Allan, V.J.; Woodman, P.G. Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat. Cell Biol. 2007, 9, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hsu, V.W.; Prekeris, R. Transport at the recycling endosome. Curr. Opin. Cell Biol. 2010, 22, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. New insights into the mechanisms of macroautophagy in mammalian cells. Internat. Rev. Cell Mol. Biol. 2008, 266, 207–247. [Google Scholar]
- Kinchen, J.M.; Ravichandran, K.S. Phagosome maturation: Going through the acid test. Nat. Rev. Mol. Cell Biol. 2008, 9, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harbor Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. A shortcut to the lysosome: The mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 2012, 107, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A.; Kauppinen, A.; Blasiak, J.; Roider, J.; Salminen, A.; Kaarniranta, K. Cellular and molecular mechanisms of age-related macular degeneration: From impaired autophagy to neovascularization. Internat. J. Biochem. Cell Biol. 2013, 45, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Lyons, J.A. Lysosomal storage diseases. J. Inborn Errors Metab. Screen. 2014, 2. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Cyphersmith, A.; Zapata, J.A.; Kim, Y.J.; Payne, C.K. Lysosome transport as a function of lysosome diameter. PloS ONE 2014, 9, e86847. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Hirokawa, N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J. Cell Biol. 1995, 131, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Wenzel, E.M.; Pedersen, N.M.; Olsvik, H.; Schink, K.O.; Schultz, S.W.; Vietri, M.; Nisi, V.; Bucci, C.; Brech, A.; et al. Repeated er-endosome contacts promote endosome translocation and neurite outgrowth. Nature 2015, 520, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; Van Der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor orp1l contacts the er protein vap to control rab7-rilp-p150 glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mtor. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.; Williams, D.S. Isolation and culture of primary mouse retinal pigmented epithelial cells. Adv. Exp. Med. Biol. 2003, 533, 347–352. [Google Scholar] [PubMed]
- Nandrot, E.F.; Anand, M.; Almeida, D.; Atabai, K.; Sheppard, D.; Finnemann, S.C. Essential role for mfg-e8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12005–12010. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, A.M.; Trost, M.; Beyaert, R.; Hoffmann, E. Patterns, receptors, and signals: Regulation of phagosome maturation. Trends Immunol. 2017, 38, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Wavre-Shapton, S.T.; Meschede, I.P.; Seabra, M.C.; Futter, C.E. Phagosome maturation during endosome interaction revealed by partial rhodopsin processing in retinal pigment epithelium. J. Cell Sci. 2014, 127, 3852–3861. [Google Scholar] [CrossRef] [PubMed]
- Garin, J.; Diez, R.; Kieffer, S.; Dermine, J.F.; Duclos, S.; Gagnon, E.; Sadoul, R.; Rondeau, C.; Desjardins, M. The phagosome proteome: Insight into phagosome functions. J. Cell Biol. 2001, 152, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.M.; Jordao, L.; Vieira, O.V. Rab10 regulates phagosome maturation and its overexpression rescues mycobacterium-containing phagosomes maturation. Traffic 2010, 11, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of rab5 and rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell. Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Kim, M.K.; Schreiber, A.D.; Grinstein, S. Role of ubiquitin and proteasomes in phagosome maturation. Mol. Biol. Cell 2005, 16, 2077–2090. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.; Bucci, C.; Vieira, O.V.; Schroer, T.A.; Grinstein, S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of rab7 and rilp. Mol. Cell. Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.K.; Eskelinen, E.L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. Lamp proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Hasler, P.; Maschalidi, S.; Lippens, C.; Castelbou, C.; Bouvet, S.; Guido, D.; Bermont, F.; Bassoy, E.Y.; Page, N.; Merkler, D.; et al. Stim1 promotes migration, phagosomal maturation and antigen cross-presentation in dendritic cells. Nat. Commun. 2017, 8, 1852. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome ph regulation and maturation in human m1 and m2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Wandelmer, J.; Reggiori, F. Amphisomes: Out of the autophagosome shadow? EMBO J. 2013, 32, 3116–3118. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. Tfeb links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. Mtorc1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of tfeb. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor tfeb links mtorc1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and tfeb. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Von Figura, K. Metachromatic leukodystrophy. In The Metabolic and Molecular Basis of Inherited Diseases, 8th ed.; McGraw Hill: New York, NY, USA, 2001; Volume 3, pp. 3695–3724. [Google Scholar]
- Hers, H. A-glucosidase deficiency in generalized glycogen-storage disease (pompe’s disease). Biochem. J. 1963, 86, 11. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Lysosomal metabolism of glycoproteins. Glycobiology 2005, 15, 1r–15r. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A. Disease pathogenesis explained by basic science: Lysosomal storage diseases as autophagocytic disorders. Int. J. Clin. Pharmaco. Ther. 2009, 47, S34–S38. [Google Scholar] [CrossRef]
- Raben, N.; Shea, L.; Hill, V.; Plotz, P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol. 2009, 453, 417–449. [Google Scholar] [PubMed]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Jin, L.-W.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-β precursor protein in neurons with niemann-pick type c defects is associated with endosomal abnormalities. Am. J. Pathol. 2004, 164, 975–985. [Google Scholar] [CrossRef]
- Nixon, R.A. Niemann-pick type c disease and alzheimer’s disease: The app-endosome connection fattens up. Am. J. Pathol. 2004, 164, 757–761. [Google Scholar] [CrossRef]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Aufschnaiter, A.; Kohler, V.; Buttner, S. Taking out the garbage: Cathepsin d and calcineurin in neurodegeneration. Neural Regen. Res. 2017, 12, 1776–1779. [Google Scholar] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Iseki, E.; Togo, T.; Yamaguchi, A.; Katsuse, O.; Katsuyama, K.; Kanzaki, S.; Shiozaki, K.; Kawanishi, C.; Yamashita, S. Neuronal and glial accumulation of α-and β-synucleins in human lipidoses. Acta Neuropathol. 2007, 114, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2007, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. A-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S. Α-synuclein impairs macroautophagy: Implications for parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Evin, G. Beta-site app-cleaving enzyme 1 trafficking and alzheimer’s disease pathogenesis. J. Neurochem. 2012, 120, 869–880. [Google Scholar] [PubMed]
- Wu, J.; Petralia, R.S.; Kurushima, H.; Patel, H.; Jung, M.Y.; Volk, L.; Chowdhury, S.; Shepherd, J.D.; Dehoff, M.; Li, Y. Arc/arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell 2011, 147, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Kant, R. Stuck in traffic: An emerging theme in diseases of the nervous system. Trends Neurosci. 2014, 37, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S.; Lee, J.H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic alzheimer’s disease and down syndrome: Differential effects of apoe genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Abeta localization in abnormal endosomes: Association with earliest abeta elevations in ad and down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.-H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O. Macroautophagy—a novel β-amyloid peptide-generating pathway activated in alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Kumar, A.; Peterhoff, C.; Kulnane, L.S.; Uchiyama, Y.; Lamb, B.; Cuervo, A.; Nixon, R. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for β-amyloid peptide over-production and localization in alzheimer’s disease. Internat. J. Biochem. Cell Biol 2004, 36, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal alzheimer aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Benzing, W.C.; Mufson, E.J.; Armstrong, D.M. Alzheimer’s disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative disease unless amyloid β-protein deposition is present. Brain Res. 1993, 606, 10–18. [Google Scholar] [CrossRef]
- Suzuki, K.; Terry, R.D. Fine structural localization of acid phosphatase in senile plaques in alzheimer’s presenile dementia. Acta Neuropathol. 1967, 8, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic alzheimer’s disease: Neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [PubMed]
- Ji, Z.-S.; Miranda, R.D.; Newhouse, Y.M.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein e4 potentiates amyloid β peptide-induced lysosomal leakage and apoptosis in neuronal cells. J. Biol. Chem. 2002, 277, 21821–21828. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of down syndrome and alzheimer’s disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Staras, K.; Branco, T.; Burden, J.J.; Pozo, K.; Darcy, K.; Marra, V.; Ratnayaka, A.; Goda, Y. A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron 2010, 66, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Ratnayaka, A.; Marra, V.; Branco, T.; Staras, K. Extrasynaptic vesicle recycling in mature hippocampal neurons. Nat. Commun. 2011, 2, 531. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ratnayaka, A.; Marra, V.; Bush, D.; Burden, J.J.; Branco, T.; Staras, K. Recruitment of resting vesicles into recycling pools supports nmda receptor-dependent synaptic potentiation in cultured hippocampal neurons. J. Physiol. 2012, 590, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–116. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2015, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L. Age and disease-related structural changes in the retinal pigment epithelium. Clin. Ophthalmol. 2008, 2, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Finnemann, S.C.; Bonilha, V.L.; Marmorstein, A.D.; Rodriguez-Boulan, E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc. Natl. Acad Sci. USA 1997, 94, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Yasumura, D.; Matthes, M.T.; LaVail, M.M.; Vollrath, D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J. Biol. Chem. 2002, 277, 17016–17022. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, P.M.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; LaVail, M.M.; Vollrath, D. Mutation of the receptor tyrosine kinase gene mertk in the retinal dystrophic rcs rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Ratnayaka, J.A.; Lynn, S.A.; Griffiths, H.; Scott, J.; Cree, A.; Lotery, A.J. An ex-vivo platform for manipulation and study of retinal pigment epithelial (rpe) cells in long-term culture. Investig. Ophthalmol. Visual Sci. 2015, 56, 2332. [Google Scholar]
- Lynn, S.A.; Ward, G.; Keeling, E.; Scott, J.A.; Cree, A.J.; Johnston, D.A.; Page, A.; Cuan-Urquizo, E.; Bhaskar, A.; Grossel, M.C.; et al. Ex-vivo models of the retinal pigment epithelium (rpe) in long-term culture faithfully recapitulate key structural and physiological features of native rpe. Tissue Cell 2017, 49, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Godino, R.; Garland, D.L.; Pierce, E.A. Isolation, culture and characterization of primary mouse rpe cells. Nat. Protoc. 2016, 11, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, F.; Safa, H.; Finnemann, S.C. Understanding photoreceptor outer segment phagocytosis: Use and utility of rpe cells in culture. Exp. Eye Res. 2014, 126, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dorey, C.K.; Wu, G.; Ebenstein, D.; Garsd, A.; Weiter, J. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Investig. Ophthalmol. Visual Sci. 1989, 30, 1691–1699. [Google Scholar]
- Krohne, T.U.; Stratmann, N.K.; Kopitz, J.; Holz, F.G. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp. Eye Res. 2010, 90, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.O.; Abrams, T. Kinetic studies of rod outer segment binding and ingestion by cultured rat rpe cells. Exp. Eye Res. 1987, 45, 907–922. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Rodriguez-Boulan, E. Macrophage and retinal pigment epithelium phagocytosis: Apoptotic cells and photoreceptors compete for alphavbeta3 and alphavbeta5 integrins, and protein kinase c regulates alphavbeta5 binding and cytoskeletal linkage. J. Exp. Med. 1999, 190, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Mayerson, P.L.; Hall, M.O. Rat retinal pigment epithelial cells show specificity of phagocytosis in vitro. J. Cell Biol. 1986, 103, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.; Kitamoto, J.; Williams, D.S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin viia, the usher syndrome 1b protein. Proc. Natl. Acad. Sci. USA 2003, 100, 6481–6486. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, T.; Lane, A.; Laughlin, W.E.; Cheetham, M.E.; Futter, C.E. Correlative light and immuno-electron microscopy of retinal tissue cryostat sections. PLoS ONE 2018, 13, e0191048. [Google Scholar] [CrossRef] [PubMed]
- Law, A.L.; Ling, Q.; Hajjar, K.A.; Futter, C.E.; Greenwood, J.; Adamson, P.; Wavre-Shapton, S.T.; Moss, S.E.; Hayes, M.J. Annexin a2 regulates phagocytosis of photoreceptor outer segments in the mouse retina. Mol. Biol. Cell 2009, 20, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Esteve-Rudd, J.; Lopes, V.S.; Diemer, T.; Lillo, C.; Rump, A.; Williams, D.S. Microtubule motors transport phagosomes in the rpe, and lack of klc1 leads to amd-like pathogenesis. J. Cell Biol. 2015, 210, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Finnemann, S.C.; Leung, L.W.; Rodriguez-Boulan, E. The lipofuscin component a2e selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 3842–3847. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Finnemann, S.C. Analysis of photoreceptor outer segment phagocytosis by rpe cells in culture. Methods Mol. Biol. 2013, 935, 285–295. [Google Scholar] [PubMed]
- Feeney-Burns, L.; Hilderbrand, E.S.; Eldridge, S. Aging human rpe: Morphometric analysis of macular, equatorial, and peripheral cells. Investig. Ophthalmol. Visual Sci 1984, 25, 195–200. [Google Scholar]
- Sundelin, S.; Wihlmark, U.; Nilsson, S.E.G.; Brunk, U.T. Lipofuscin accumulation in cultured retinal pigment epithelial cells reduces their phagocytic capacity. Curr. Eye Res. 1998, 17, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Sitte, N.; Huber, M.; Grune, T.; Ladhoff, A.; Doecke, W.-d.; Von Zglinicki, T.; Davies, K.J. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. 2000, 14, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Soura, V.; Stewart-Parker, M.; Williams, T.L.; Ratnayaka, A.; Atherton, J.; Gorringe, K.; Tuffin, J.; Darwent, E.; Rambaran, R.; Klein, W.; et al. Visualization of co-localization in abeta42-administered neuroblastoma cells reveals lysosome damage and autophagosome accumulation related to cell death. Biochem. J. 2012, 441, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Moriarty, P.; Jarvis-Evans, J.; Marcyniuk, B. Regional variation and age-related changes of lysosomal enzymes in the human retinal pigment epithelium. Br. J. Ophthalmol. 1994, 78, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, F.A.; Boulton, M.E. Inhibition of rpe lysosomal and antioxidant activity by the age pigment lipofuscin. Investig. Ophthalmol. Visual Sci. 2001, 42, 3041–3046. [Google Scholar]
- Brunk, U.; Wihlmark, U.; Wrigstad, A.; Roberg, K.; Nilsson, S. Accumulation of lipofuscin within retinal pigment epithelial cells results in enhanced sensitivity to photo-oxidation. Gerontology 1995, 41, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Parish, C.A.; Hashimoto, M.; Nakanishi, K.; Dillon, J.; Sparrow, J. Isolation and one-step preparation of a2e and iso-a2e, fluorophores from human retinal pigment epithelium. Proc. Natl. Acad.Sci. USA 1998, 95, 14609–14613. [Google Scholar] [CrossRef] [PubMed]
- Eldred, G.E. Age pigment structure. Nature 1993, 364, 396. [Google Scholar] [CrossRef] [PubMed]
- Poliakov, E.; Strunnikova, N.V.; Jiang, J.-k.; Martinez, B.; Parikh, T.; Lakkaraju, A.; Thomas, C.; Brooks, B.P.; Redmond, T.M. Multiple a2e treatments lead to melanization of rod outer segment–challenged arpe-19 cells. Mol. Vis. 2014, 20, 285. [Google Scholar] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Katz, M.L. Incomplete proteolysis may contribute to lipofuscin accumulation in the retinal pigment epithelium. Adv. Exp. Med. Biol. 1989, 266, 109–116; discussion 116–118. [Google Scholar] [PubMed]
- Kaemmerer, E.; Schutt, F.; Krohne, T.U.; Holz, F.G.; Kopitz, J. Effects of lipid peroxidation-related protein modifications on rpe lysosomal functions and pos phagocytosis. Investig. Ophthalmol. Visual Sci. 2007, 48, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the rpe is associated with increased susceptibility to oxidative stress and amd. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Schubert, T.; Gleiser, C.; Heiduschka, P.; Franz, C.; Nagel-Wolfrum, K.; Sahaboglu, A.; Weisschuh, N.; Eske, G.; Rohbock, K.; Rieger, N.; et al. Deletion of myosin vi causes slow retinal optic neuropathy and age-related macular degeneration (amd)-relevant retinal phenotype. Cell. Mol. Life Sci. 2015, 72, 3953–3969. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy receptors link myosin vi to autophagosomes to mediate tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Salminen, A.; Eskelinen, E.-L.; Kopitz, J. Heat shock proteins as gatekeepers of proteolytic pathways—implications for age-related macular degeneration (amd). Ageing Res. Rev. 2009, 8, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. Mtorc1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar h+-atpase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mtorc1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. Zkscan3 is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Liu, J.; Baltazar, G.; Laties, A.M.; Mitchell, C.H. Rescue of compromised lysosomes enhances degradation of photoreceptor outer segments and reduces lipofuscin-like autofluorescence in retinal pigmented epithelial cells. Adv. Exp. Med. Biol. 2014, 801, 105–111. [Google Scholar] [PubMed]
- Pfeffer, B.A.; Philp, N.J. Cell culture of retinal pigment epithelium: Special issue. Exp. Eye Res. 2014, 126, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.R.G.; Brown, F.E.; Cree, A.J.; Ratnayaka, J.A.; Lotery, A.J. Sorsby fundus dystrophy—A review of pathology and disease mechanisms. Exp. Eye Res. 2017, 165, 35–46. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keeling, E.; Lotery, A.J.; Tumbarello, D.A.; Ratnayaka, J.A. Impaired Cargo Clearance in the Retinal Pigment Epithelium (RPE) Underlies Irreversible Blinding Diseases. Cells 2018, 7, 16. https://doi.org/10.3390/cells7020016
Keeling E, Lotery AJ, Tumbarello DA, Ratnayaka JA. Impaired Cargo Clearance in the Retinal Pigment Epithelium (RPE) Underlies Irreversible Blinding Diseases. Cells. 2018; 7(2):16. https://doi.org/10.3390/cells7020016
Chicago/Turabian StyleKeeling, Eloise, Andrew J. Lotery, David A. Tumbarello, and J. Arjuna Ratnayaka. 2018. "Impaired Cargo Clearance in the Retinal Pigment Epithelium (RPE) Underlies Irreversible Blinding Diseases" Cells 7, no. 2: 16. https://doi.org/10.3390/cells7020016
APA StyleKeeling, E., Lotery, A. J., Tumbarello, D. A., & Ratnayaka, J. A. (2018). Impaired Cargo Clearance in the Retinal Pigment Epithelium (RPE) Underlies Irreversible Blinding Diseases. Cells, 7(2), 16. https://doi.org/10.3390/cells7020016