Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress




Abstract
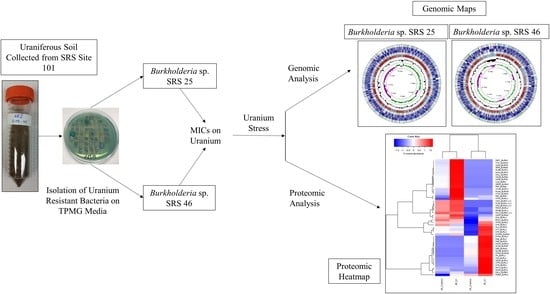
1. Introduction
2. Material and Methods
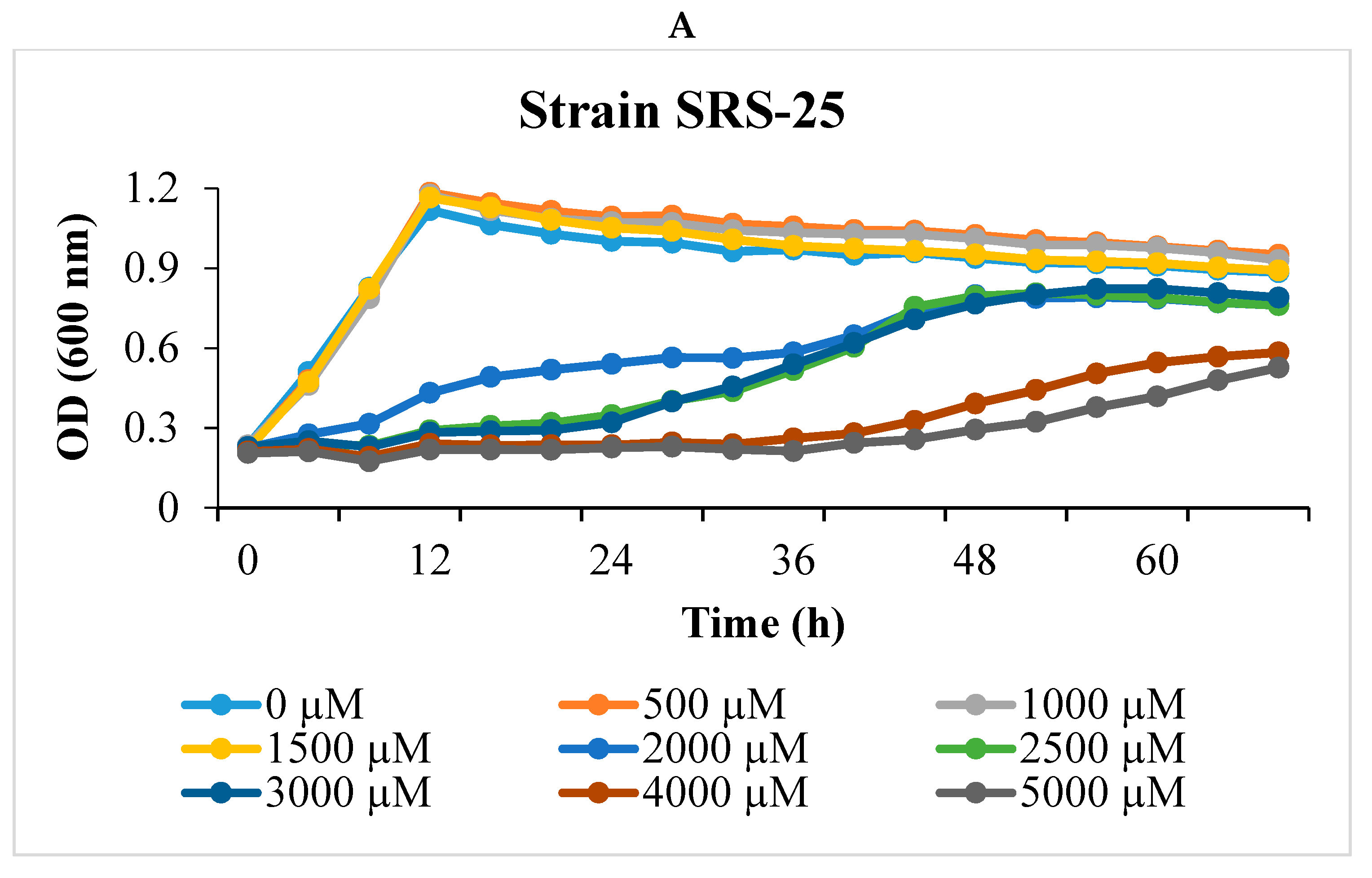
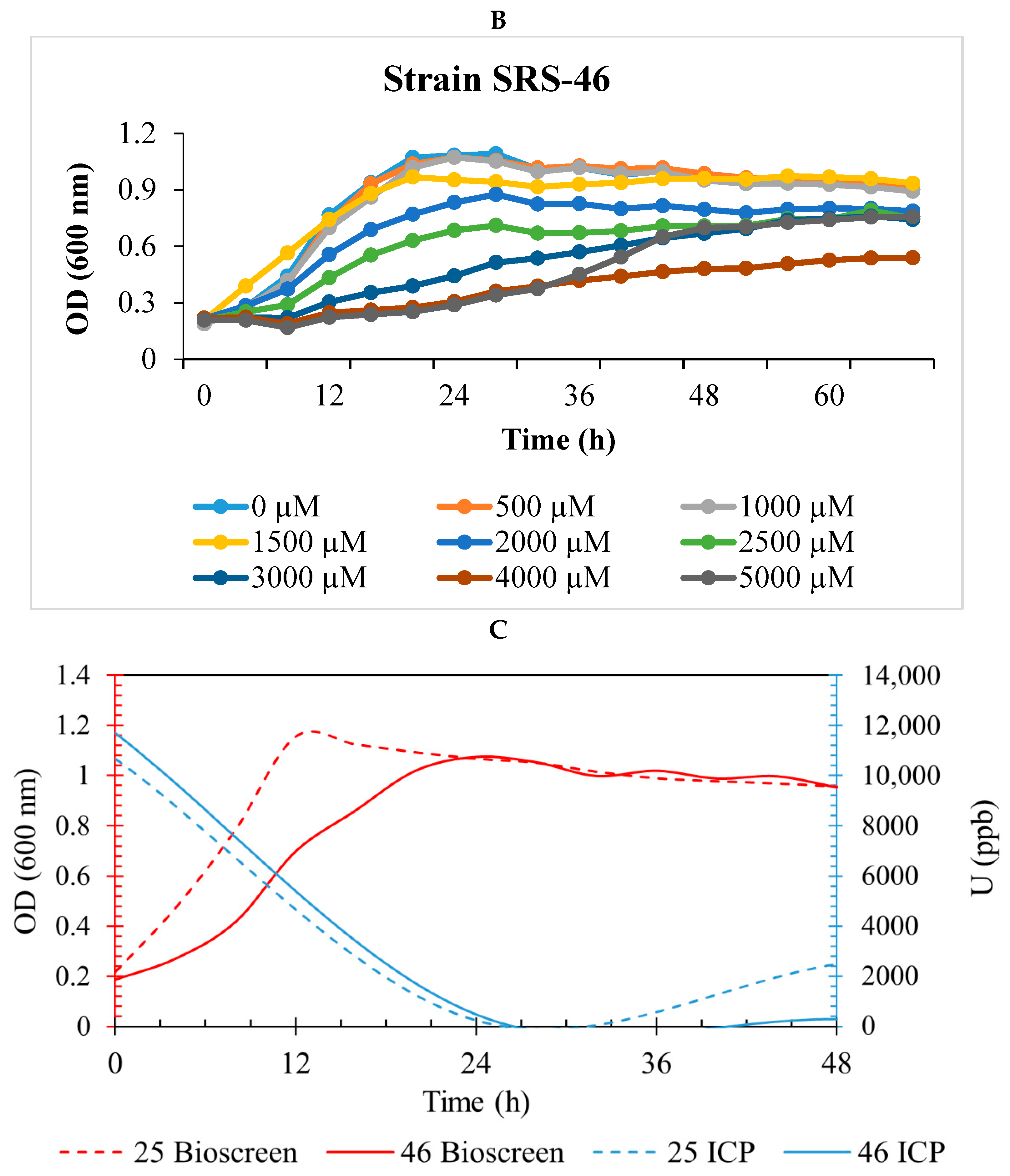
2.1. Isolation and U Resistance Studies on Strains SRS-25 and SRS-46
2.2. Uranium Depletion by Strains SRS-25 and SRS-46
2.3. Genomic Characterization of Strains SRS-25 and SRS-46
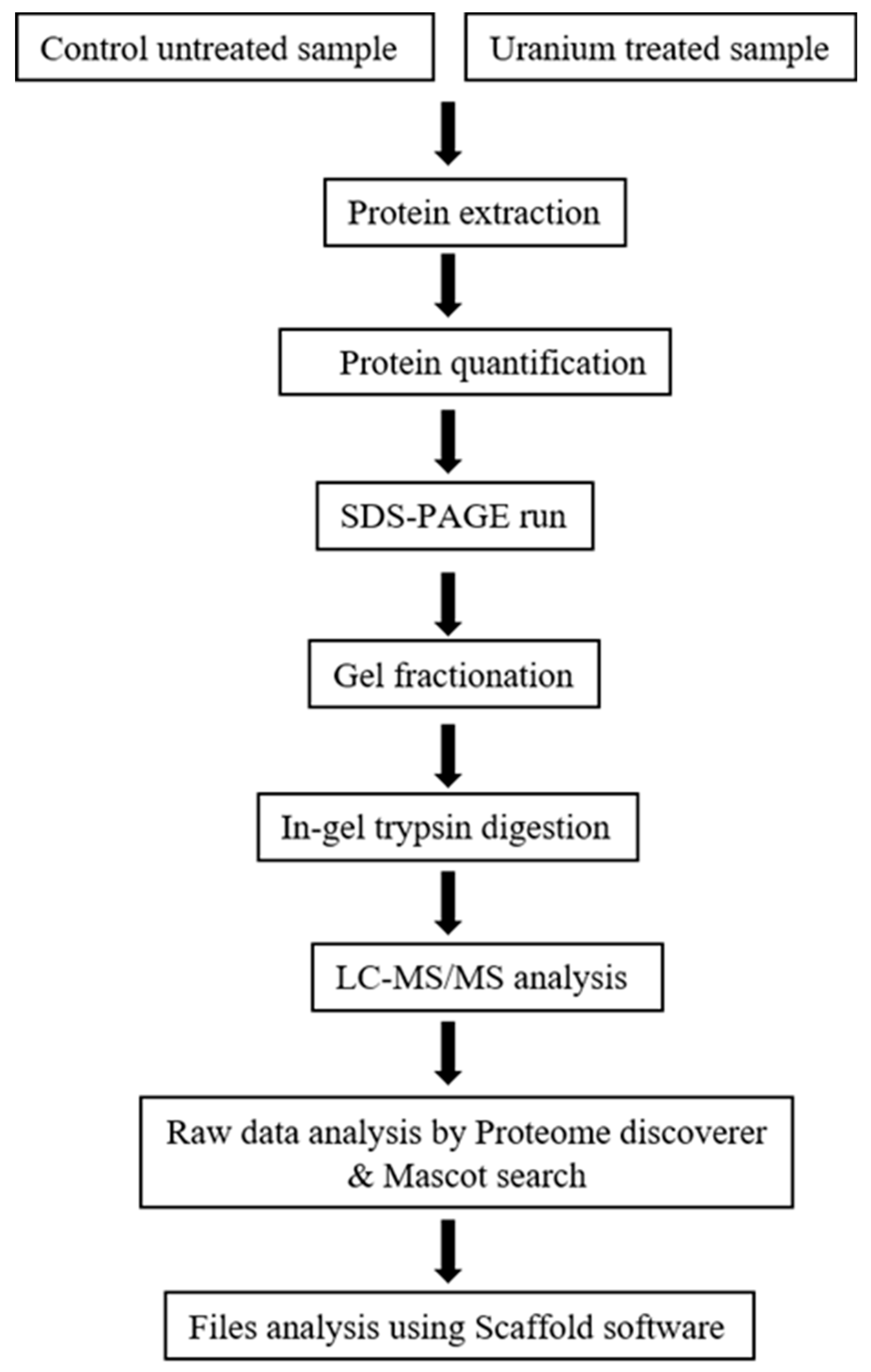
2.4. Protein Extraction and Separation
2.5. Mass Spectrometry and Protein Identification
2.6. Statistical Analysis
2.7. Genomic and Proteomic Data Accession Numbers
3. Results
3.1. Depletion of Uranium by Strains SRS-25 and SRS-46
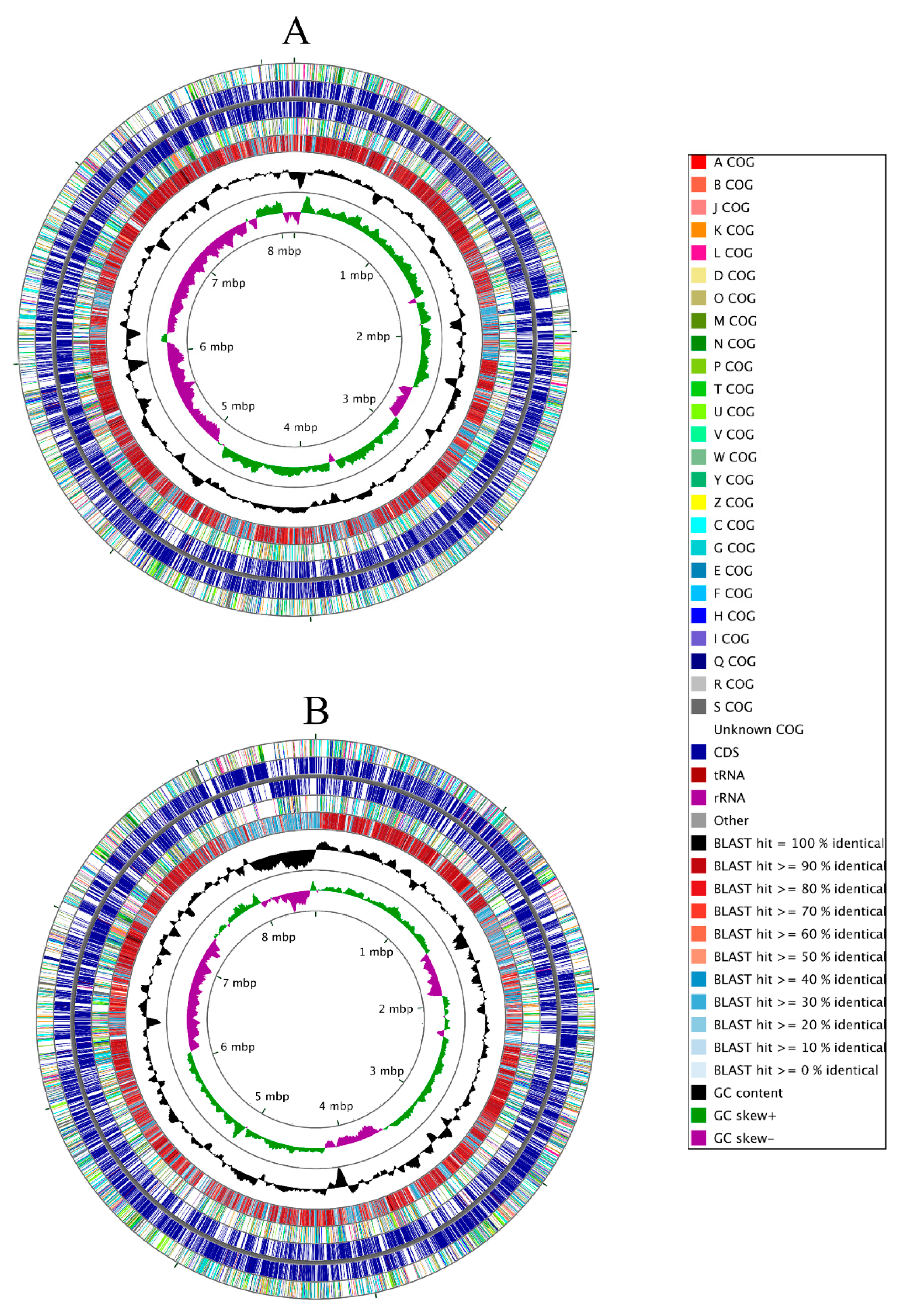
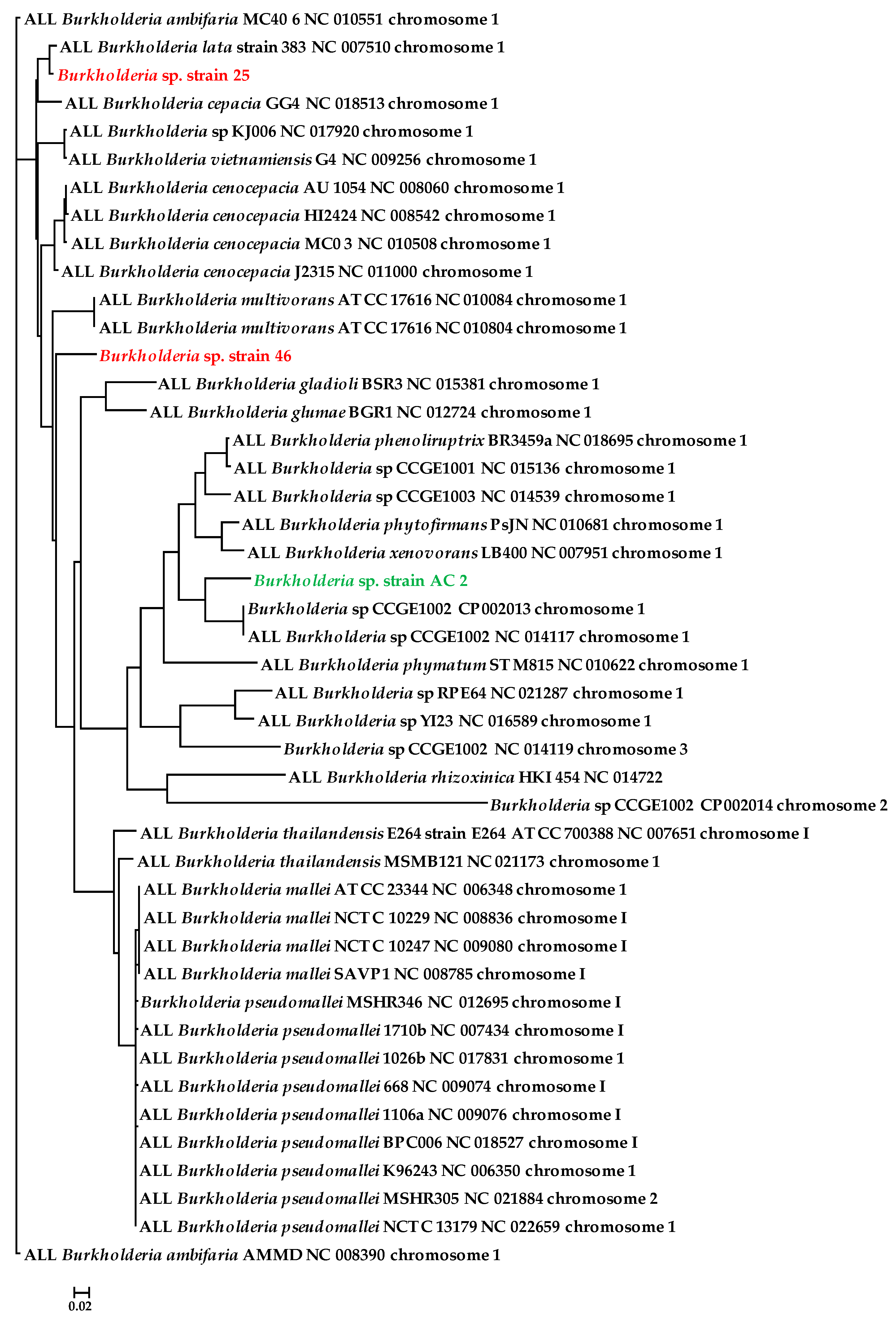
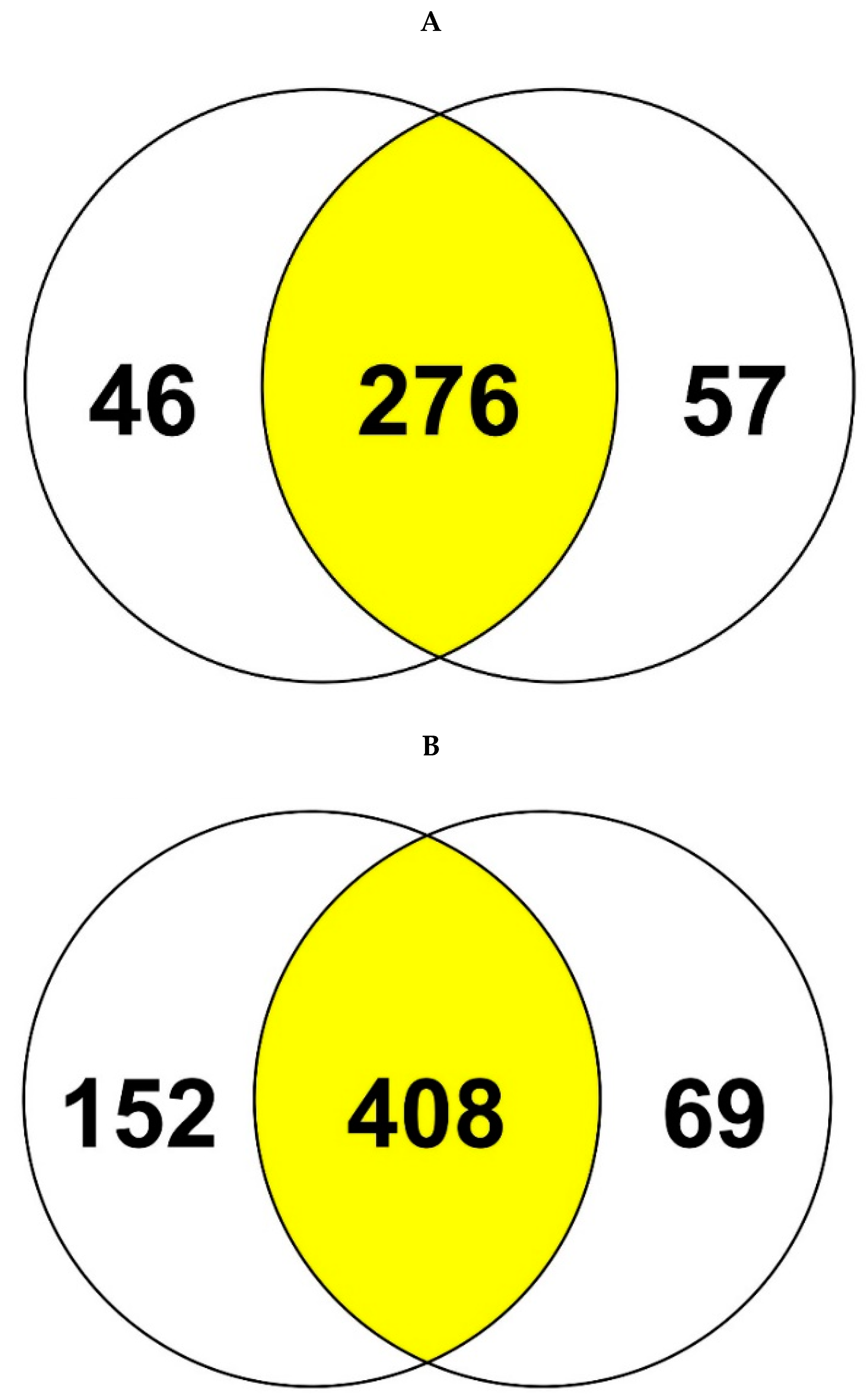
3.2. Genome-Centric Evaluation to Identify Metal Resistance Basis of Isolated Strains
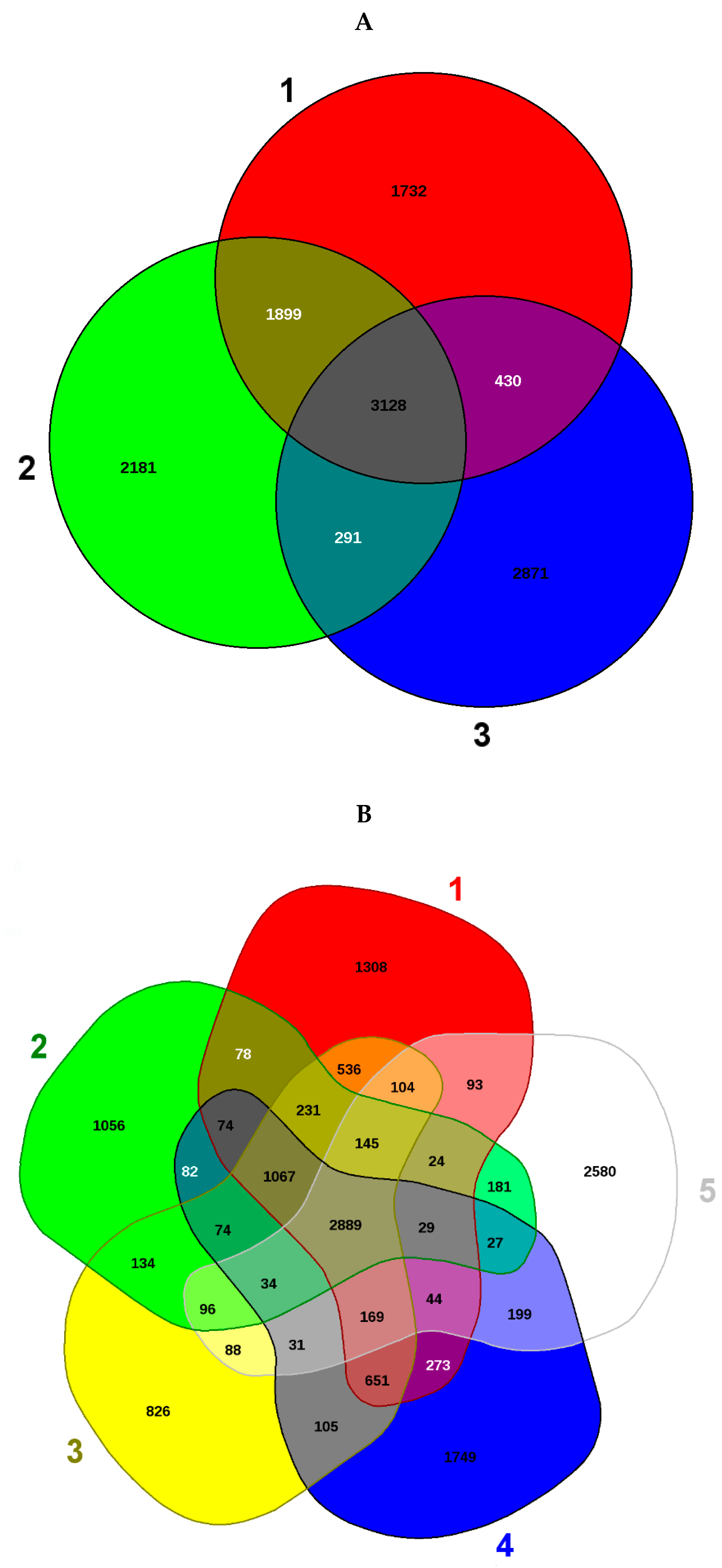
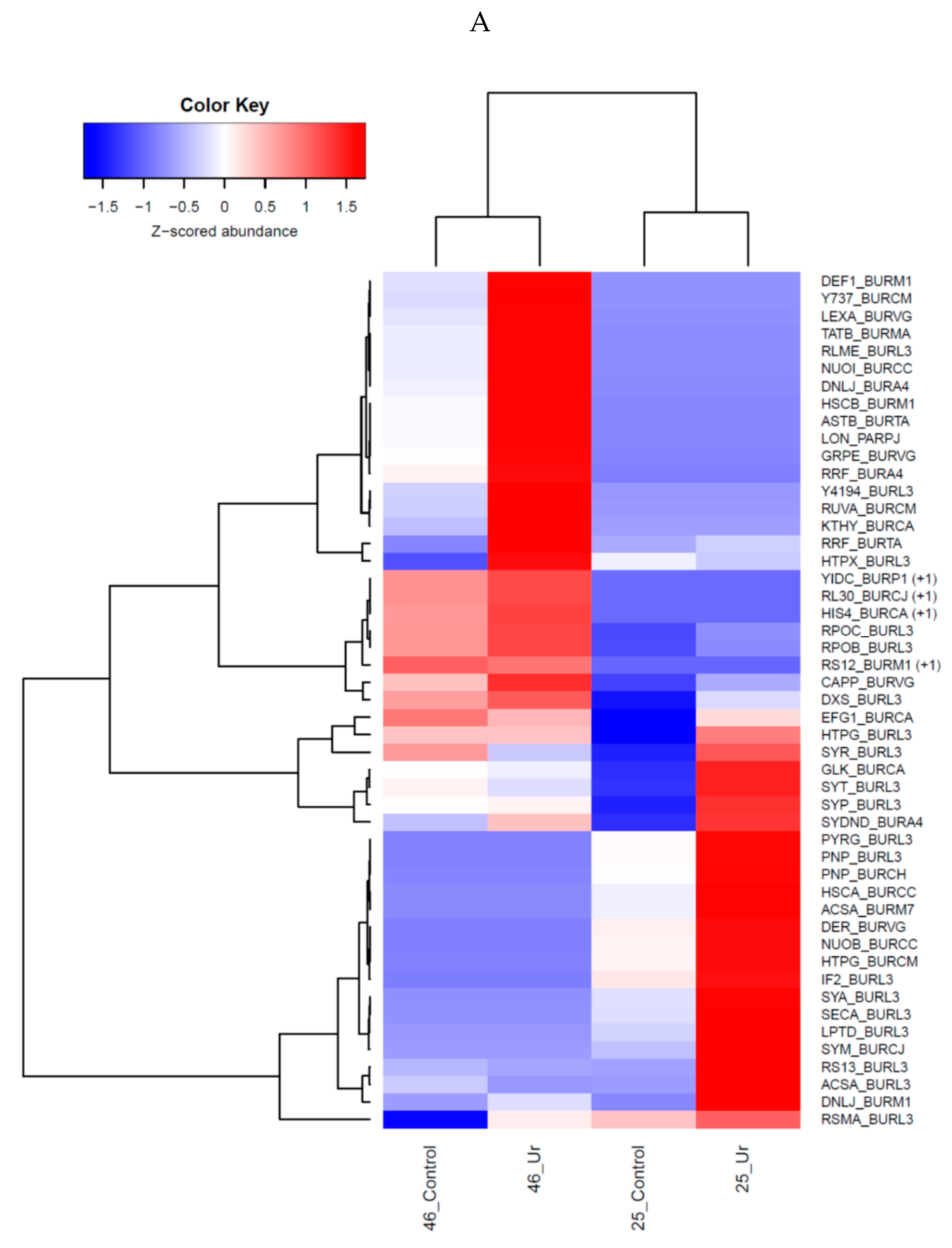
3.3. Comparative Proteomics Study to Analyze Response to Uranium Exposure
3.4. Protein Biosynthesis and Growth
3.5. DNA Damage, Repair and Stress Response
3.6. Metabolism and ATP Synthesis
3.7. Membrane Damage and Other Proteins
3.8. Uncharacterized Proteins
3.9. Statistical Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riley, R.G.; Zachara, J.M. Chemical Contaminants on DOE Lands and Selection of Contaminant Mixtures for Subsurface Science Research; Pacific Northwest Laboratory: Richland, WA, USA, 1992.
- Evans, A.; Bauer, L.; Haselow, J.; Hayes, D.; Martin, H.; McDowell, W.; Pickett, J. Uranium in the Savannah River Site Environment; Westinghouse Savannah River Co.: Aiken, SC, USA, 1992. [Google Scholar]
- Martinez, R.J.; Wu, C.H.; Beazley, M.J.; Andersen, G.L.; Conrad, M.E.; Hazen, T.C.; Taillefert, M.; Sobecky, P.A. Microbial community responses to organophosphate substrate additions in contaminated subsurface sediments. PLoS ONE 2014, 9, e100383. [Google Scholar] [CrossRef] [PubMed]
- Newsome, L.; Morris, K.; Lloyd, J.R. The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chem. Geol. 2014, 363, 164–184. [Google Scholar] [CrossRef]
- Ye, Q. Microbial Diversity Associated with Metal- and Radionuclide-Contamination at the DOE Savannah River Site (SRS), South Carolina, USA. Ph.D. Thesis, East China Normal University, Shanghai, China, 2007. [Google Scholar]
- Kumar, R.; Nongkhlaw, M.; Acharya, C.; Joshi, S.R. Uranium (U)-tolerant bacterial diversity from U ore deposit of Domiasiat in North-East India and its prospective utilisation in bioremediation. Microbes Environ. 2013, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Misra, C.S.; Gupta, A.; Ballal, A.; Apte, S.K. Interaction of uranium with bacterial cell surfaces: Inferences from phosphatase-mediated uranium precipitation. Appl. Environ. Microbiol. 2016, 82, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.J.; Beazley, M.J.; Taillefert, M.; Arakaki, A.K.; Skolnick, J.; Sobecky, P.A. Aerobic uranium (VI) bioprecipitation by metal-resistant bacteria isolated from radionuclide- and metal-contaminated subsurface soils. Environ. Microbiol. 2007, 9, 3122–3133. [Google Scholar] [CrossRef] [PubMed]
- Beazley, M.J.; Martinez, R.J.; Sobecky, P.A.; Webb, S.M.; Taillefert, M. Uranium biomineralization as a result of bacterial phosphatase activity: Insights from bacterial isolates from a contaminated subsurface. Environ. Sci. Technol. 2007, 41, 5701–5707. [Google Scholar] [CrossRef]
- Mumtaz, S.; Streten-Joyce, C.; Parry, D.L.; McGuinness, K.A.; Lu, P.; Gibb, K.S. Fungi outcompete bacteria under increased uranium concentration in culture media. J. Environ. Radioact. 2013, 120, 39–44. [Google Scholar] [CrossRef]
- Guo, H.; Luo, S.; Chen, L.; Xiao, X.; Xi, Q.; Wei, W.; Zeng, G.; Liu, C.; Wan, Y.; Chen, J.; et al. Bioremediation of heavy metals by growing hyperaccumulaor endophytic bacterium Bacillus sp. L14. Bioresour. Technol. 2010, 101, 8599–8605. [Google Scholar] [CrossRef]
- Das, S.; Dash, H.R.; Chakraborty, J. Genetic basis and importance of metal resistant genes in bacteria for bioremediation of contaminated environments with toxic metal pollutants. Appl. Microbiol. Biotechnol. 2016, 100, 2967–2984. [Google Scholar] [CrossRef]
- Pathak, A.; Chauhan, A.; Stothard, P.; Green, S.; Maienschein-Cline, M.; Jaswal, R.; Seaman, J. Genome-centric evaluation of Burkholderia sp. strain SRS-W-2-2016 resistant to high concentrations of uranium and nickel isolated from the Savannah River Site (SRS), USA. Genom. Data 2017, 12, 62–68. [Google Scholar] [CrossRef]
- Suzuki, Y.; Banfield, J.F. Resistance to, and accumulation of, uranium by bacteria from a uranium-contaminated site. Geomicrobiol. J. 2004, 21, 113–121. [Google Scholar] [CrossRef]
- North, N.N.; Dollhopf, S.L.; Petrie, L.; Istok, J.D.; Balkwill, D.L.; Kostka, J.E. Change in bacterial community structure during in situ biostimulation of subsurface sediment cocontaminated with uranium and nitrate. Appl. Environ. Microbiol. 2004, 70, 4911–4920. [Google Scholar] [CrossRef] [PubMed]
- Koribanics, N.M.; Tuorto, S.J.; Lopez-Chiaffarelli, N.; McGuinness, L.R.; Häggblom, M.M.; Williams, K.H.; Long, P.E.; Kerkhof, L.J. Spatial distribution of an uranium-respiring betaproteobacterium at the Rifle, CO field research site. PLoS ONE 2015, 10, e0123378. [Google Scholar] [CrossRef] [PubMed]
- Lessie, T.G.; Hendrickson, W.; Manning, B.D.; Devereux, R. Genomic complexity and plasticity of Burkholderia cepacia. FEMS Microbiol. Lett. 1996, 144, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Gallois, N.; Alpha-Bazin, B.; Ortet, P.; Barakat, M.; Piette, L.; Long, J.; Berthomieu, C.; Armengaud, J.; Chapon, V. Proteogenomic insights into uranium tolerance of a Chernobyl’s Microbacterium bacterial isolate. J. Proteom. 2018, 177, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.C.; Ma, J.; Salemi, M.R.; Phinney, B.S.; Bowman, G.R.; Jiao, Y. Shotgun proteomic analysis unveils survival and detoxification strategies by Caulobacter crescentus during exposure to uranium, chromium, and cadmium. J. Proteome Res. 2014, 13, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Brodie, E.L.; Suzuki, Y.; McAdams, H.H.; Andersen, G.L. Whole-genome transcriptional analysis of heavy metal stresses in Caulobacter crescentus. J. Bacteriol. 2005, 187, 8437–8449. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Pathak, A.; Jaswal, R.; Edwards, B., III; Chappell, D.; Ball, C.; Garcia-Sillas, R.; Stothard, P.; Seaman, J. Physiological and comparative genomic analysis of arthrobacter sp. SRS-W-1-2016 provides insights on niche adaptation for survival in uraniferous soils. Genes 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Riccio, M.L.; Rossolini, G.M.; Lombardi, G.; Chiesurin, A.; Satta, G. Expression cloning of different bacterial phosphatase-encoding genes by histochemical screening of genomic libraries onto an indicator medium containing phenolphthalein diphosphate and methyl green. J. Appl. Microbiol. 1997, 82, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, J.D.; Khijniak, T.J.; Neely, B.; Sattar, M.A.; Sowder, A.G.; Mills, G.; Bertsch, P.M.; Morris, P.J. Reduction of nickel and uranium toxicity and enhanced trichloroethylene degradation to Burkholderia vietnamiensis PR1301 with hydroxyapatite amendment. Environ. Sci. Technol. 2007, 41, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9. [Google Scholar] [CrossRef]
- Minot, S.S.; Krumm, N.; Greenfield, N.B. One codex: A sensitive and accurate data platform for genomic microbial identification. BioRxiv 2015. [Google Scholar] [CrossRef]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.-J.; Zakrzewski, M.; Goesmann, A. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 2009, 10, 154. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Pinto-Carbó, M.; Sieber, S.; Dessein, S.; Wicker, T.; Verstraete, B.; Gademann, K.; Eberl, L.; Carlier, A. Evidence of horizontal gene transfer between obligate leaf nodule symbionts. ISME J. 2016, 10, 2092–2105. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Urban, T.A.; Goldberg, J.B. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Eberl, L.; Vandamme, P. Members of the genus burkholderia: Good and bad guys. F1000Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Orellana, R.; Hixson, K.K.; Murphy, S.; Mester, T.; Sharma, M.L.; Lipton, M.S.; Lovley, D.R. Proteome of geobacter sulfurreducens in the presence of U(VI). Microbiology 2014, 160, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, E.; Nakajima, A. Effect of catechins and tannins on depleted uranium-induced DNA strand breaks. J. Radioanal. Nucl. Chem. 2012, 293, 711–714. [Google Scholar] [CrossRef]
- Zobel, C.R.; Beer, M. Electron stains. I. Chemical studies on the interaction of DNA with uranyl salts. J. Biophys. Biochem. Cytol. 1961, 10, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.A. Microbial molecular chaperones. Adv. Microb. Physiol. 2001, 44, 93–140. [Google Scholar] [PubMed]
- Merroun, M.L.; Selenska-Pobell, S. Bacterial interactions with uranium: An environmental perspective. J. Contam. Hydrol. 2008, 102, 285–295. [Google Scholar] [CrossRef]
- Morcillo, F.; González-Muñoz, M.T.; Reitz, T.; Romero-González, M.E.; Arias, J.M.; Merroun, M.L. Biosorption and biomineralization of U(VI) by the marine bacterium Idiomarina loihiensis MAH1: Effect of background electrolyte and pH. PLoS ONE 2014, 9, e91305. [Google Scholar] [CrossRef]
- Dekker, L.; Arsène-Ploetze, F.; Santini, J.M. Comparative proteomics of Acidithiobacillus ferrooxidans grown in the presence and absence of uranium. Res. Microbiol. 2016, 167, 234–239. [Google Scholar] [CrossRef]
- Panda, B.; Basu, B.; Acharya, C.; Rajaram, H.; Apte, S.K. Proteomic analysis reveals contrasting stress response to uranium in two nitrogen-fixing Anabaena strains, differentially tolerant to uranium. Aquat. Toxicol. 2017, 182, 205–213. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Homologue |
|---|---|
| Transporter proteins | Proline/Betaine transporter |
| MFS-type transporter YhjX, YcaD | |
| Phospholipid ABC transporter permease protein | |
| ABC transporter ATP-binding protein YbhF, YheS | |
| Cystine inner membrane transporter | |
| Hemolysin transporter protein ShlB precursor | |
| Divalent metal cation transporter MntH | |
| Inner membrane ABC transporter permease protein | |
| Riboflavin transporter | |
| Dicarboxylic acid transporter DauA | |
| Inner membrane transporter yiJE, YedA, YnfM | |
| Sulfoacetate transporter SauU | |
| Citrate transporter | |
| Lysophospholipid transporter LplT | |
| Niacin/nicotinamide transporter NaiP | |
| (3-hydroxy-phenyl) propionate transporter | |
| Tartrate transporter | |
| H(+)/Cl(−) exchange transporter ClcA | |
| d-galactonate transporter | |
| Low-affinity inorganic phosphate transporter 1 | |
| l-galactonate (Hexuronate) transporter | |
| Heme/hemopexin transporter protein HuxB precursor | |
| Sialic acid transporter | |
| Amino-acid permease protein YxeN | |
| Glutamate/aspartate transporter permease protein | |
| Fluoride ion transporter CrcB | |
| Sialic acid transporter | |
| Fe (3+) ions import ATP-binding protein FbpC | |
| 4-hydroxybenzoate transporter PcaK | |
| Glucarate transporter | |
| Efflux pump membrane transporter BepE | |
| Nitrate/nitrite transporter NarK2 | |
| Lactose transport system permease protein LacF | |
| Manganese ABC transporter substrate-binding lipoprotein | |
| Uric acid transporter UacT | |
| Glutamine ABC transporter permease protein GlnM | |
| High-affinity gluconate transporter | |
| Phospholipid ABC transporter-binding protein MlaD | |
| Stress proteins | Stress response kinase |
| General stress protein 39, 69 | |
| Universal stress protein | |
| Persistence and stress-resistance antitoxin PasI | |
| Persistence and stress-resistance toxin PasT | |
| TRAP-T-associated universal stress protein TeaD | |
| Acid stress protein IbaG | |
| Cytochromes | Cytochrome c oxidase subunit 2 precursor |
| Cytochrome c oxidase subunit 1 | |
| Cytochrome c oxidase assembly protein CtaG | |
| Cytochrome c oxidase subunit 3 | |
| Cytochrome bd ubiquinol oxidase subunit X | |
| Cytochrome bd-I ubiquinol oxidase subunit 2 | |
| Cytochrome bd ubiquinol oxidase subunit 1 | |
| Gluconate 2-dehydrogenase cytochrome c subunit precursor | |
| Cytochrome c4 precursor | |
| Cytochrome b561 | |
| Succinate dehydrogenase cytochrome b556 subunit | |
| Cytochrome c-552 precursor | |
| Cytochrome c-554(548) | |
| Quinone-reactive Ni/Fe-hydrogenase B-type cytochrome subunit | |
| Fructose dehydrogenase cytochrome subunit precursor | |
| Sulfide dehydrogenase [flavocytochrome c] flavoprotein chain precursor | |
| Cytochrome bo (3) ubiquinol oxidase subunit 4 | |
| Cytochrome bo (3) ubiquinol oxidase subunit 3 | |
| Cytochrome bo (3) ubiquinol oxidase subunit 2 precursor | |
| Cytochrome c biogenesis protein CcsA | |
| Cytochrome c1 precursor | |
| Cytochrome b/c1 | |
| Ubiquinol-cytochrome c reductase iron-sulfur | |
| Cytochrome c biogenesis protein CcsB | |
| Cytochrome c-555 precursor | |
| Cytochrome b556(fdo) subunit | |
| Metal resistance proteins | ATM1-type heavy metal exporter |
| Divalent metal cation transporter MntH | |
| ATP-dependent zinc metalloprotease FtsH | |
| Metal-dependent hydrolase YcfH, YjjV | |
| Metallo-beta-lactamase type 2 | |
| Metal chaperone YciC | |
| Metallo-hydrolase YycJ | |
| Metalloprotease PmbA | |
| High-affinity nickel transport protein | |
| Nickel and cobalt resistance protein CnrA | |
| Nickel-binding periplasmic protein | |
| Magnesium and cobalt efflux protein CorC | |
| Cobalt-zinc-cadmium resistance protein CzcA, CzcC, CzcB | |
| Cobalt/magnesium transport protein CorA | |
| Copper resistance protein C precursor | |
| Copper-exporting P-type ATPase A | |
| Copper homeostasis protein CutC | |
| Arsenical resistance operon repressor | |
| Arsenical-resistance protein Acr3 | |
| Manganese ABC transporter substrate-binding lipoprotein | |
| Manganese transport system membrane protein MntB | |
| ATP-dependent zinc metalloprotease FtsH | |
| Zinc-type alcohol dehydrogenase-like protein | |
| Zinc uptake regulation protein | |
| Zinc import ATP-binding protein ZnuC | |
| High-affinity zinc uptake system membrane protein ZnuB | |
| Zinc transport protein ZntB | |
| Cadmium-transporting ATPase | |
| Drug resistance | Multidrug resistance protein MdtH, MdtE, MdtC, NorM, stp |
| Multidrug export protein EmrB, EmrA | |
| Multidrug resistance outer membrane protein MdtP | |
| Multidrug efflux pump subunit AcrB | |
| Multidrug resistance protein 3, EmrK | |
| Multidrug export ATP-binding/permease protein |
| Function | Protein | Gene | Fold Change | Strain |
|---|---|---|---|---|
| Protein synthesis, translation and transport | Aspartate-tRNA(Asp/Asn) ligase | aspS | 2.3 | SRS-25 |
| Alanine-tRNA ligase | alaS | 4.5 | SRS-25 | |
| Elongation factor G1 | fusA1 | 2.4 | SRS-25 | |
| Proline-tRNA ligase | proS | 5.6 | SRS-25 | |
| Arginine-tRNA ligase | argS | 3.8 | SRS-25 | |
| Glutamate-tRNA ligase | gltX | 2 | SRS-25 | |
| Methionine-tRNA ligase | metG | 9.6 | SRS-25 | |
| Translation initiation factor IF-2 | infB | 2.3 | SRS-25 | |
| Elongation factor G2 | fusA2 | 2.1 | SRS-25 | |
| Protein translocase subunit SecA | secA | 4.7 | SRS-25 | |
| Threonine-tRNA ligase | thrS | 5.2 | SRS-25 | |
| 50S ribosomal protein L20 | rplT | 1.9 | SRS-25 | |
| 50S ribosomal protein L10 | rplJ | 1.7 | SRS-25 | |
| 30S ribosomal protein S13 | rpsM | 3.2 | SRS-25 | |
| 30S ribosomal protein S7 | rpsG | 1.9 | SRS-46 | |
| 30S ribosomal protein S5 | rpsE | 1.8 | SRS-46 | |
| 50S ribosomal protein L5 | rplE | 1.8 | SRS-46 | |
| Ribosome-recycling factor | frr | 2.7 | SRS-46 | |
| 50S ribosomal protein L25 | rplY | 1.8 | SRS-46 | |
| NADH-quinone oxidoreductase subunit C | nuoC | 2 | SRS-46 | |
| Sec-independent protein translocase protein tatB | tatB | 3.8 | SRS-46 | |
| Peptide deformylase 1 | def1 | 4.7 | SRS-46 | |
| Ribosomal RNA small subunit methyltransferase A | rsmA | 3.8 | SRS-46 | |
| Ribosomal RNA large subunit methyltransferase E | rlmE | 3.8 | SRS-46 | |
| Electron transport coupled proton transport | NADH-quinone oxidoreductase subunit B | nuoB | 2.5 | SRS-25 |
| Ribosome biogenesis | GTPase Der | der | 2.5 | SRS-25 |
| 30S ribosomal protein S13 | rpsM | 3.2 | SRS-25 | |
| Endoribonuclease YbeY | ybeY | 2.5 | SRS-46 | |
| Amino acid biosynthesis | Dihydroxy-acid dehydratase 1 | ilvD1 | 2.3 | SRS-25 |
| Argininosuccinate lyase | argH | 1.8 | SRS-46 | |
| Imidazole glycerol phosphate synthase subunit HisF | hisF | 1.7 | SRS-46 | |
| 3-isopropylmalate dehydratase small subunit | leuD | 1.8 | SRS-46 | |
| Homoserine O-succinyltransferase | metXS | 1.7 | SRS-46 | |
| Imidazole glycerol phosphate synthase subunit HisH | hisH | 2.5 | SRS-46 | |
| Transcription | DNA-directed RNA polymerase subunit beta | rpoB | 3.8 | SRS-25 |
| DNA-directed RNA polymerase subunit beta | rpoC | 2.9 | SRS-25 | |
| Bifunctional protein glk | glk | 3.1 | SRS-25 | |
| DNA Replication, recombination and repair | Chaperone protein DnaK | dnaK | 1.9 | SRS-25 |
| DNA ligase | ligA | 17 | SRS-25 | |
| DNA ligase | ligA | 2.5 | SRS-46 | |
| lexA repressor | lexA | 4.2 | SRS-46 | |
| Holiday junction ATP-dependent DNA helicase RuvA | ruvA | 7 | SRS-46 | |
| Stress response | Chaperone protein HtpG | htpG | 2.6 | SRS-25 |
| Chaperone protein HscA homolog | hscA | 3.5 | SRS-25 | |
| Polyribonucleotide nucleotidyltransferase | pnp | 2.9 | SRS-25 | |
| N-succinylglutamate 5-semialdehyde dehydrogenase | astD | 2.4 | SRS-25 | |
| Lon protease | lon | 3.2 | SRS-46 | |
| Protease HtpC homolog | htpX | 3.1 | SRS-46 | |
| Protein GrpE | grpE | 3 | SRS-46 | |
| Co-chaperone protein HscB homolog | hscB | 3.2 | SRS-46 | |
| 60 kDa chaperonin 2 | groL2 | 1.7 | SRS-46 | |
| Chaperone protein DnaJ | dnaJ | 1.7 | SRS-46 | |
| Heat-inducible transcription repressor HrcA | hrcA | 1.9 | SRS-46 | |
| Nucleotide biosynthesis | CTP synthase | pyrG | 2.8 | SRS-25 |
| Thymidylate kinase | tmk | 11 | SRS-46 | |
| Isoprene and thiamine biosynthesis | 1-deoxy-D-xylulose-5-phosphate synthase | dxs | 5.4 | SRS-25 |
| 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase | ispF | 2.1 | SRS-25 | |
| CO2 fixation | Phosphoenolpyruvate carboxylase | ppc | 3.6 | SRS-25 |
| Phosphoenolpyruvate carboxylase | ppc | 1.9 | SRS-46 | |
| Outer membrane assembly protein, lipopolysaccharide transport | LPS-assembly protein LptD | lptD | 5.8 | SRS-25 |
| Oxidative stress response and protein repair | Peptide methionine sulfoxide reductase MsrB | msrB | 1.9 | SRS-25 |
| Metabolism | Acetyl-coenzyme A synthetase | acsA | 3.6 | SRS-25 |
| Enolase | eno | 1.7 | SRS-46 | |
| N-succinylarginine dihydrolase | astB | 3.2 | SRS-46 | |
| Thymidine phosphorylase | deoA | 2.5 | SRS-46 | |
| Phophoglucosamine mutase | glmM | 1.7 | SRS-46 | |
| Anhydro-N-acetylmuramic acid kinase | anmk | 2.5 | SRS-46 | |
| dCTP deaminase | dcd | 1.7 | SRS-46 | |
| Nucleotide biosynthesis | Thymidylate kinase | tmk | 11 | SRS-46 |
| Oxidoreductase activity and antibiotic response | Thiol:disulfide interchange protein DsbA | dsbA | 2.2 | SRS-46 |
| Ion channnel, ion transport | Large-conductance mechanosensitive channel | mscL | 2.5 | SRS-46 |
| Aerobic respiration, ATP synthesis | NADH-quinone oxidoreductase subunit 1 | nuol | 3.8 | SRS-46 |
| ATP synthase subunit delta | atpH | 1.9 | SRS-46 | |
| Kinase, transferase | Cytidylate kinase | cmk | 1.7 | SRS-46 |
| Lipid biosynthesis | 3-hydroxy-[acyl-carrier-protein] | fabZ | 1.9 | SRS-46 |
| Lipid-A-disaccharide synthase | lpxB | 1.9 | SRS-46 | |
| Pyridoxine biosynthesis | 4-hydroxythreonine-4-phosphate dehydrogenase | pdxA | 2.5 | SRS-46 |
| Queuosine biosynthesis | 7-carboxy-7-deazaguanine synthase | queE | 2.5 | SRS-46 |
| Ubiquinone biosynthesis | 2-nonaprenyl-3-methyl-6-methoxy-1,4 benzoquinol hydroxylase | coq7 | 1.7 | SRS-46 |
| Glycosyl transferase | Uracil phosphoribosyltransferase | upp | 2.1 | SRS-46 |
| Poorly characterized | UPF0234 protein Bmul_0741/BMUL1_02519 | Bmul_0741 | 1.9 | SRS-46 |
| UPF0307 protein Bcep18194_A4194 | Bcep18194_A4194 | 6.4 | SRS-46 | |
| Probable transcriptional regulatory protein Bphyt_1301 | Bphyt_1301 | 2.1 | SRS-46 | |
| UPF0234 protein Bphy_0527 | Bphy_0527 | 1.7 | SRS-46 | |
| Probable transcriptional regulatory protein Bamb_2332 | Bamb_2332 | 2.3 | SRS-46 | |
| UPF0301 protein Bamb_0737 | Bamb_0737 | 5.1 | SRS-46 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, M.; Pathak, A.; Rathore, R.S.; Prakash, O.; Singh, R.; Jaswal, R.; Seaman, J.; Chauhan, A. Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress. Cells 2018, 7, 269. https://doi.org/10.3390/cells7120269
Agarwal M, Pathak A, Rathore RS, Prakash O, Singh R, Jaswal R, Seaman J, Chauhan A. Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress. Cells. 2018; 7(12):269. https://doi.org/10.3390/cells7120269
Chicago/Turabian StyleAgarwal, Meenakshi, Ashish Pathak, Rajesh Singh Rathore, Om Prakash, Rakesh Singh, Rajneesh Jaswal, John Seaman, and Ashvini Chauhan. 2018. "Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress" Cells 7, no. 12: 269. https://doi.org/10.3390/cells7120269
APA StyleAgarwal, M., Pathak, A., Rathore, R. S., Prakash, O., Singh, R., Jaswal, R., Seaman, J., & Chauhan, A. (2018). Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress. Cells, 7(12), 269. https://doi.org/10.3390/cells7120269