Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy



Abstract
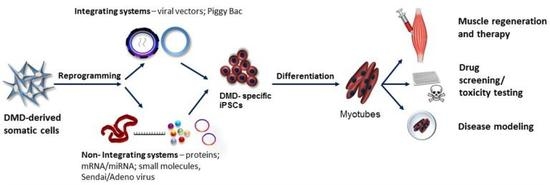
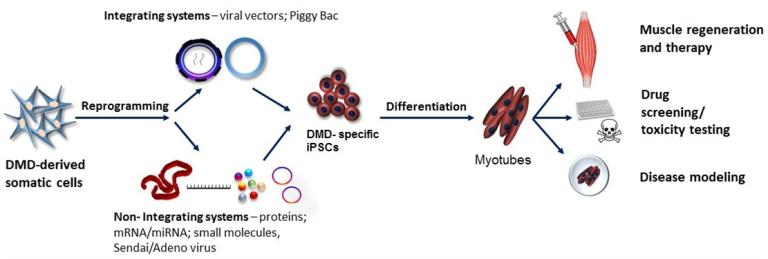
1. Introduction
2. iPSCs Generation Techniques
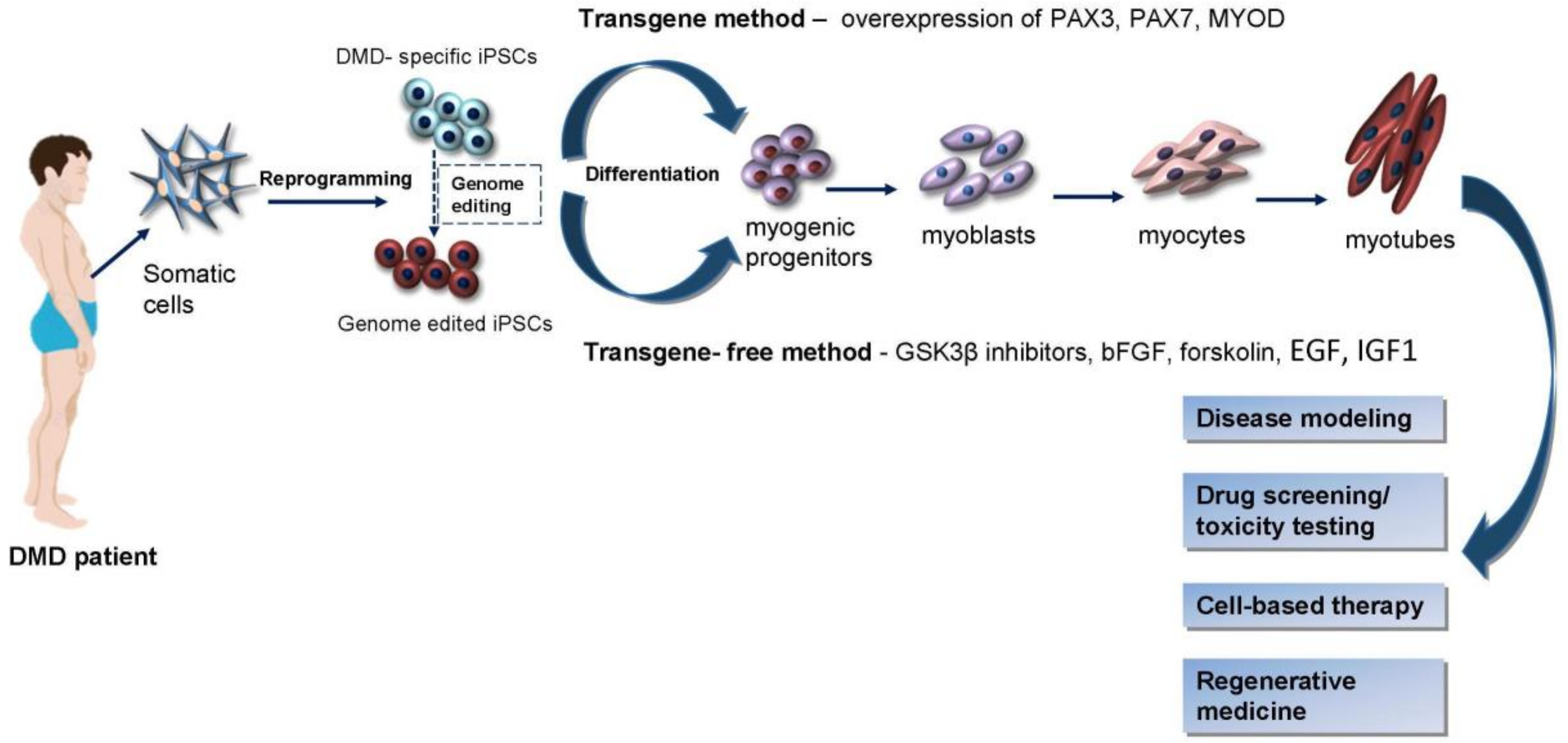
3. Induction of Myogenic Progenitors and Precursor Cells from iPSCs
4. iPSCs in Duchenne Muscular Dystrophy Modeling
5. Genetic Correction
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Rubis, P.; Wisniowska-Smialek, S.; Tomkiewicz-Pajak, L.; Kudlinski, D.; Olszowska, M.; Podolec, P. Severe course of dilated cardiomyopathy associated with Duchenne muscular dystrophy. JRCD 2014, 2, 18–22. [Google Scholar]
- Mavrogeni, S.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Kolovou, G. Cardiac involvement in Duchenne and Becker muscular dystrophy. World J. Cardiol. 2015, 7, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Beytía, M.L.; Vry, J.; Kirschner, J. Drug treatment of Duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol. 2012, 31, 4–8. [Google Scholar]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef]
- Lu, Q.-L.; Yokota, T.; Takeda, S.; Garcia, L.; Muntoni, F.; Partridge, T. The status of exon skipping as a therapeutic approach to Duchenne muscular dystrophy. Mol. Ther. 2011, 19, 9–15. [Google Scholar] [CrossRef]
- Brussee, V.; Tardif, F.; Roy, B.; Goulet, M.; Sebille, A.; Tremblay, J.P. Successful myoblast transplantation in fibrotic muscles: No increased impairment by the connective tissue1. Transplantation 1999, 67, 1618–1622. [Google Scholar] [CrossRef]
- Skuk, D.; Goulet, M.; Roy, B.; Piette, V.; Côté, C.H.; Chapdelaine, P.; Hogrel, J.-Y.; Paradis, M.; Bouchard, J.-P.; Sylvain, M.; et al. First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: Eighteen months follow-up. Neuromuscul. Disord. 2007, 17, 38–46. [Google Scholar] [CrossRef]
- Mouly, V.; Aamiri, A.; Périé, S.; Mamchaoui, K.; Barani, A.; Bigot, A.; Bouazza, B.; François, V.; Furling, D.; Jacquemin, V.; et al. Myoblast transfer therapy: is there any light at the end of the tunnel? Acta Myol. 2005, 24, 128–133. [Google Scholar] [PubMed]
- Zamborsky, R.; Rusnakova, J.; Nicodemou, A. Tissue engineering of articular cartilage: A mini-review. OnLine J. Biological. Sci. 2014, 14, 248–253. [Google Scholar] [CrossRef]
- Ostrovidov, S.; Shi, X.; Sadeghian, R.B.; Salehi, S.; Fujie, T.; Bae, H.; Ramalingam, M.; Khademhosseini, A. Stem cell differentiation toward the myogenic lineage for muscle tissue regeneration: A focus on muscular dystrophy. Stem Cell Rev. 2015, 11, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Shin, K.; Lee, S.B.; Lee, D.R.; Kwon, H. Patient-tailored application for duchene muscular dystrophy on mdx mice based induced mesenchymal stem cells. Exp. Mol. Pathol. 2014, 97, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Fan, Y.; Chen, X.; Yue, L.; Yu, B.; Li, Q.; Chen, Y.; Sun, X. Modeling induced pluripotent stem cells from fibroblasts of Duchenne muscular dystrophy patients. Int. J. Neurosci. 2014, 124, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human ipsc derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (ipscs) by temperature-sensitive sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of mir-302 and mir-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef]
- Goudenege, S.; Lebel, C.; Huot, N.B.; Dufour, C.; Fujii, I.; Gekas, J.; Rousseau, J.; Tremblay, J.P. Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol. Ther. 2012, 20, 2153–2167. [Google Scholar] [CrossRef]
- Behringer, R.; Gertsenstein, M.; Nagy, K.V.; Nagy, A. Reprogramming mouse fibroblasts with piggyBac transposons. Cold Spring Harb. Protoc. 2017, 2017. [Google Scholar] [CrossRef]
- Baranek, M.; Markiewicz, W.; Barciszewski, J. Selected small molecules as inducers of pluripotency. Acta Biochim. Pol. 2016, 63, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Schöler, H.R.; Ding, S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by oct4 and klf4 with small-molecule compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only oct4 and sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Pan, W.; Bosnakovski, D.; Baik, J.; Kyba, M.; Perlingeiro, R.C. Functional myogenic engraftment from mouse iPS cells. Stem Cell Rev. 2011, 7, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.T.; Davis, J.; Lee, G.; Mack, D.L.; Kim, D.H. Muscular dystrophy in a dish: Engineered human skeletal muscle mimetics for disease modeling and drug discovery. Drug Discov. Today 2016, 21, 1387–1398. [Google Scholar] [CrossRef]
- Dekel, I.; Magal, Y.; Pearson-White, S.; Emerson, C.P.; Shani, M. Conditional conversion of ES cells to skeletal muscle by an exogenous MYOD1 gene. New Biol. 1992, 4, 217–224. [Google Scholar]
- Darabi, R.; Gehlbach, K.; Bachoo, R.M.; Kamath, S.; Osawa, M.; Kamm, K.E.; Kyba, M.; Perlingeiro, R.C. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat. Med. 2008, 14, 134–143. [Google Scholar] [CrossRef]
- Mizuno, Y.; Chang, H.; Umeda, K.; Niwa, A.; Iwasa, T.; Awaya, T.; Fukada, S.; Yamamoto, H.; Yamanaka, S.; Nakahata, T.; et al. Generation of skeletal muscle stem/progenitor cells from murine induced pluripotent stem cells. FASEB J. 2010, 24, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Gerli, M.F.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012, 4, 140. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and reproducible myogenic differentiation from human ips cells: Prospects for modeling miyoshi myopathy in vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Yasuno, T.; Osafune, K.; Sakurai, H.; Asaka, I.; Tanaka, A.; Yamaguchi, S.; Yamada, K.; Hitomi, H.; Arai, S.; Kurose, Y.; et al. Functional analysis of iPSC-derived myocytes from a patient with carnitine palmitoyltransferase II deficiency. Biochem. Biophys. Res. Commun. 2014, 448, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by talen and crispr-cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tabebordbar, M.; Iovino, S.; Ciarlo, C.; Liu, J.; Castiglioni, A.; Price, E.; Liu, M.; Barton, E.R.; Kahn, C.R.; et al. A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell 2013, 155, 909–921. [Google Scholar] [CrossRef]
- Hosoyama, T.; McGivern, J.V.; Van Dyke, J.M.; Ebert, A.D.; Suzuki, M. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl. Med. 2014, 3, 564–574. [Google Scholar] [CrossRef]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and facs-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Stem Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef]
- Van der Wal, E.; Herrero-Hernandez, P.; Wan, R.; Broeders, M.; In’t Groen, S.L.M.; Van Gestel, T.J.M.; Van IJcken, W.F.J.; Cheung, T.H.; Van der Ploeg, A.T.; Schaaf, G.J.; et al. Large-scale expansion of human iPSC-derived skeletal muscle cells for disease modeling and cell-based therapeutic strategies. Stem Cell Rep. 2018, 10, 1975–1990. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.P.; Kim, Y.J.; Kim, H.; et al. Concordant but varied phenotypes among Duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Jiwlawat, S.; Lynch, E.; Glaser, J.; Smit-Oistad, I.; Jeffrey, J.; Van Dyke, J.M.; Suzuki, M. Differentiation and sarcomere formation in skeletal myocytes directly prepared from human induced pluripotent stem cells using a sphere-based culture. Differentiation 2017, 96, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Jiwlawat, N.; Lynch, E.; Jeffrey, J.; Van Dyke, J.M.; Suzuki, M. Current progress and challenges for skeletal muscle differentiation from human pluripotent stem cells using transgene-free approaches. Stem Cells Int. 2018, 2018, 6241681. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.-P.; Stanford, W.L.; Skerjanc, I.S. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Roca, I.; Requena, J.; Edel, M.J.; Alvarez-Palomo, A.B. Myogenic precursors from iPS cells for skeletal muscle cell replacement therapy. J. Clin. Med. 2015, 4, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, H.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Perez-Pinera, P.; Brown, M.T.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 2015, 23, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single crispr-cas9 deletion strategy that targets the majority of dmd patients restores dystrophin function in hipsc-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, B.; Iyombe-Engembe, J.P.; Rousseau, J.; Tremblay, J.P.; Ouellet, D.L. From gRNA Identification to the restoration of dystrophin expression: A dystrophin gene correction strategy for duchenne muscular dystrophy mutations using the crispr-induced deletion method. Methods Mol. Biol. 2018, 1687, 267–283. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
| Integrating Systems | Non-Integrating Systems |
|---|---|
| Retroviruses [17,18] | Adenoviruses [19,20] |
| Lentiviruses [21] | Sendai virus [20,22] |
| piggyBac transposons [23] | Plasmids [24] |
| Episomal vectors [25] | |
| mRNA [26,27,28] | |
| miRNA [29] | |
| Proteins/small molecules [30,31,32] |
| Donor Cell Type | Cell Culture Method | Transgenes of Myogenic Cells | Differentiated Cell Type | Reference |
|---|---|---|---|---|
| Fibroblasts | 2D culture | MYOD-ERT | MYOD1-expressing mesangioblasts | [42] |
| Fibroblasts | EB culture | PAX7 | Myogenic precursors | [37] |
| Fibroblasts | 2D culture | MYOD | Myocytes | [43] |
| Fibroblasts | 2D culture | MYOD | Myotubes | [16] |
| Fibroblasts | 2D culture | MYOD | Myocytes | [44] |
| Fibroblasts | 2D culture | MYOD | Skeletal muscle fibers | [45] |
| Fibroblasts | 2D culture | MYOD | Myocytes | [46] |
| Fibroblasts | EB culture | MYOD | Myoblasts | [29] |
| Fibroblasts | EB culture | MYOD | Myogenic cells | [26] |
| Donor Cell Type | Cell Culture Method | Factors | Differentiated Cell Type | Reference |
|---|---|---|---|---|
| Fibroblasts | EB culture | GSK3β inhibitor, bFGF, forskolin | Myotubes | [47] |
| Fibroblasts | EZ spheres culture | bFGF-2, EGF | Myotubes | [48] |
| Fibroblasts | 2D culture | GSK3β inhibitor, bFGF | Myoblasts | [49] |
| Fibroblasts | EB culture | FGF-2, GSK3β inhibitor (CHIR99021) | Myofibers | [50] |
| Fibroblasts | 2D culture | GSK3β inhibitor (CHIR99021), DAPT | Myoblasts | [51] |
| Fibroblasts | 2D culture | GSK3β inhibitor, BMP inhibitor, bFGF, HGF, IGF1 | Myogenic progenitors | [52] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danisovic, L.; Culenova, M.; Csobonyeiova, M. Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy. Cells 2018, 7, 253. https://doi.org/10.3390/cells7120253
Danisovic L, Culenova M, Csobonyeiova M. Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy. Cells. 2018; 7(12):253. https://doi.org/10.3390/cells7120253
Chicago/Turabian StyleDanisovic, Lubos, Martina Culenova, and Maria Csobonyeiova. 2018. "Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy" Cells 7, no. 12: 253. https://doi.org/10.3390/cells7120253
APA StyleDanisovic, L., Culenova, M., & Csobonyeiova, M. (2018). Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy. Cells, 7(12), 253. https://doi.org/10.3390/cells7120253