Mechanisms of TQ-6, a Novel Ruthenium-Derivative Compound, against Lipopolysaccharide-Induced In Vitro Macrophage Activation and Liver Injury in Experimental Mice: The Crucial Role of p38 MAPK and NF-κB Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. TQ-6 Synthesis and RAW 264.7 Cell Cultivation
2.3. Cell Viability Assay
2.4. Determination of Nitric Oxide Production
2.5. Separation of Cytoplasmic and Nuclear Extracts
2.6. Immunofluorescence Staining Assay
2.7. LPS-Induced Acute Liver Inflammation in Mice
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
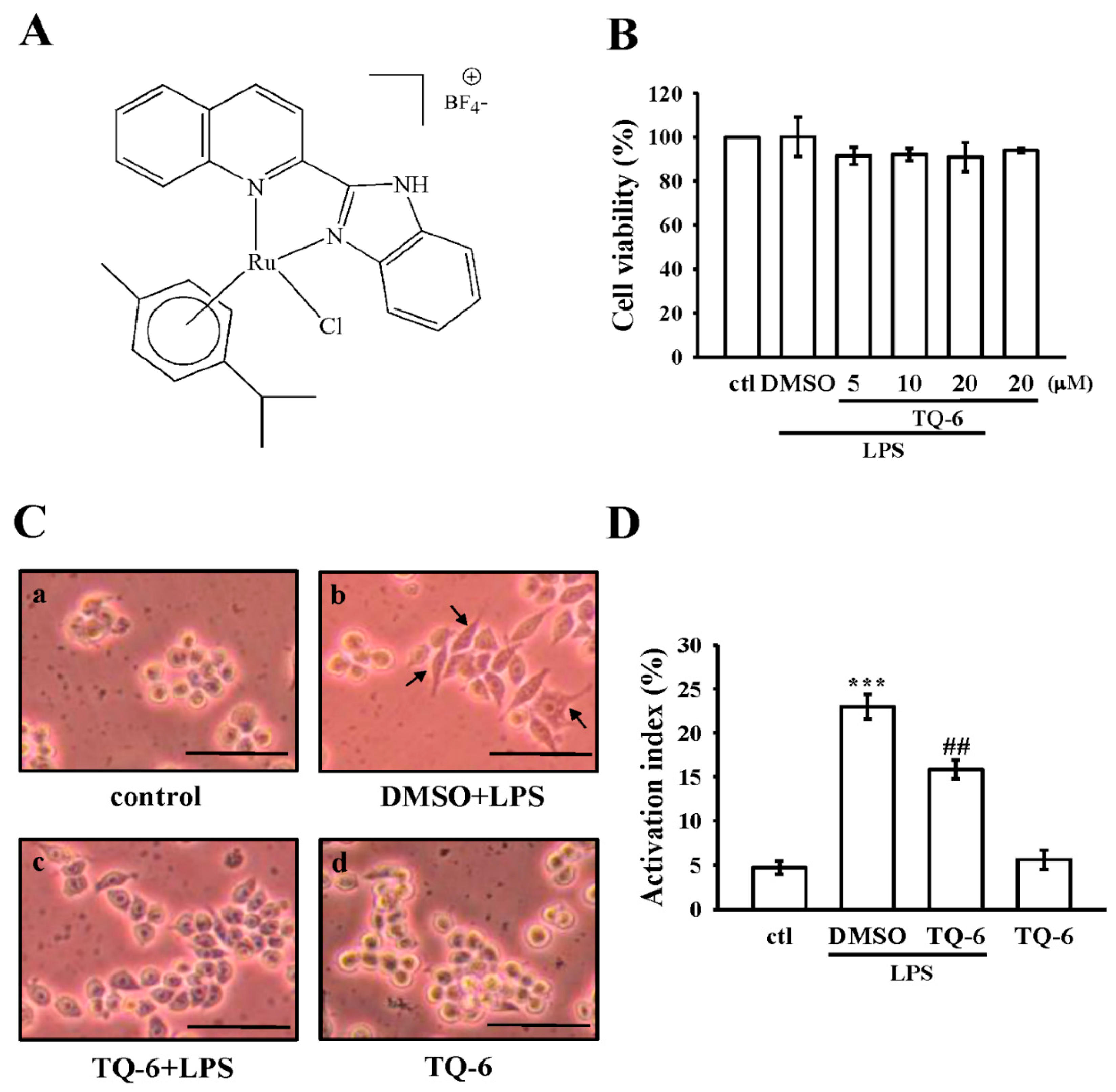
3.1. Effects of TQ-6 on Cytotoxicity and Morphology in RAW 264.7 Cells
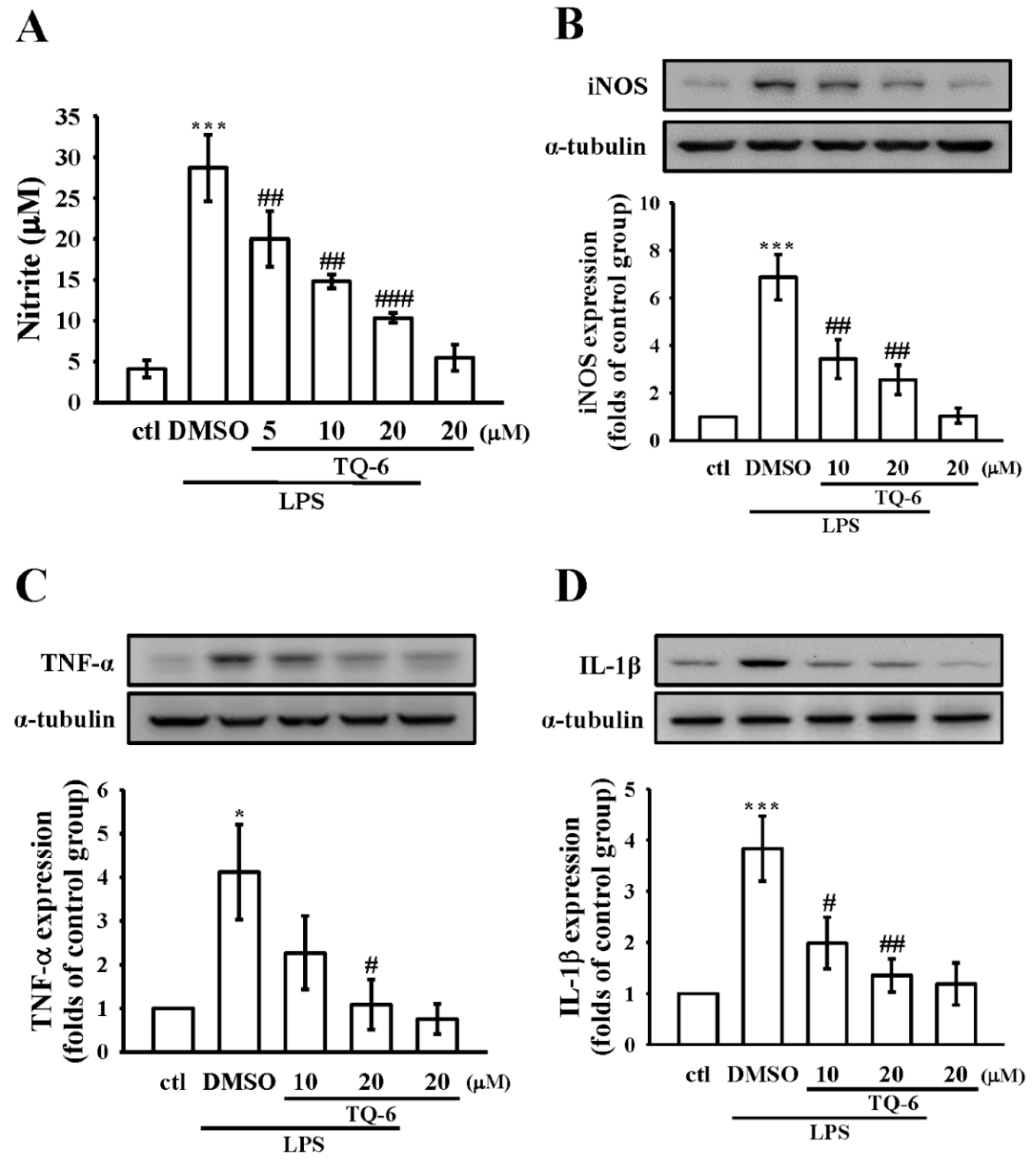
3.2. Effect of TQ-6 on NO Production and iNOS Expression in RAW Cells
3.3. Effect of TQ-6 on Pro-Inflammatory Cytokines Expression in RAW Cells
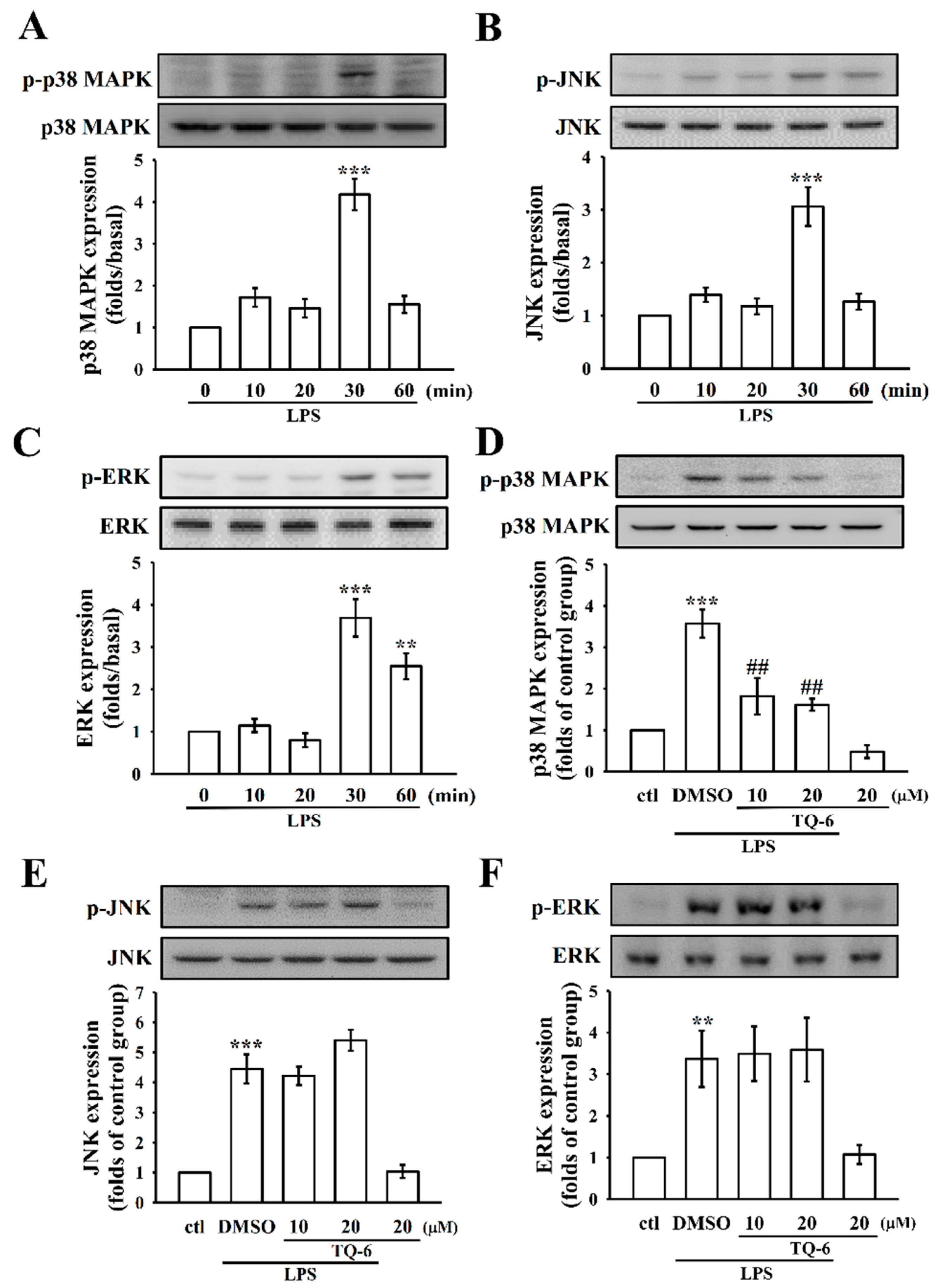
3.4. The Influence of TQ-6 on MAPKs in LPS-Induced RAW Cell Activation
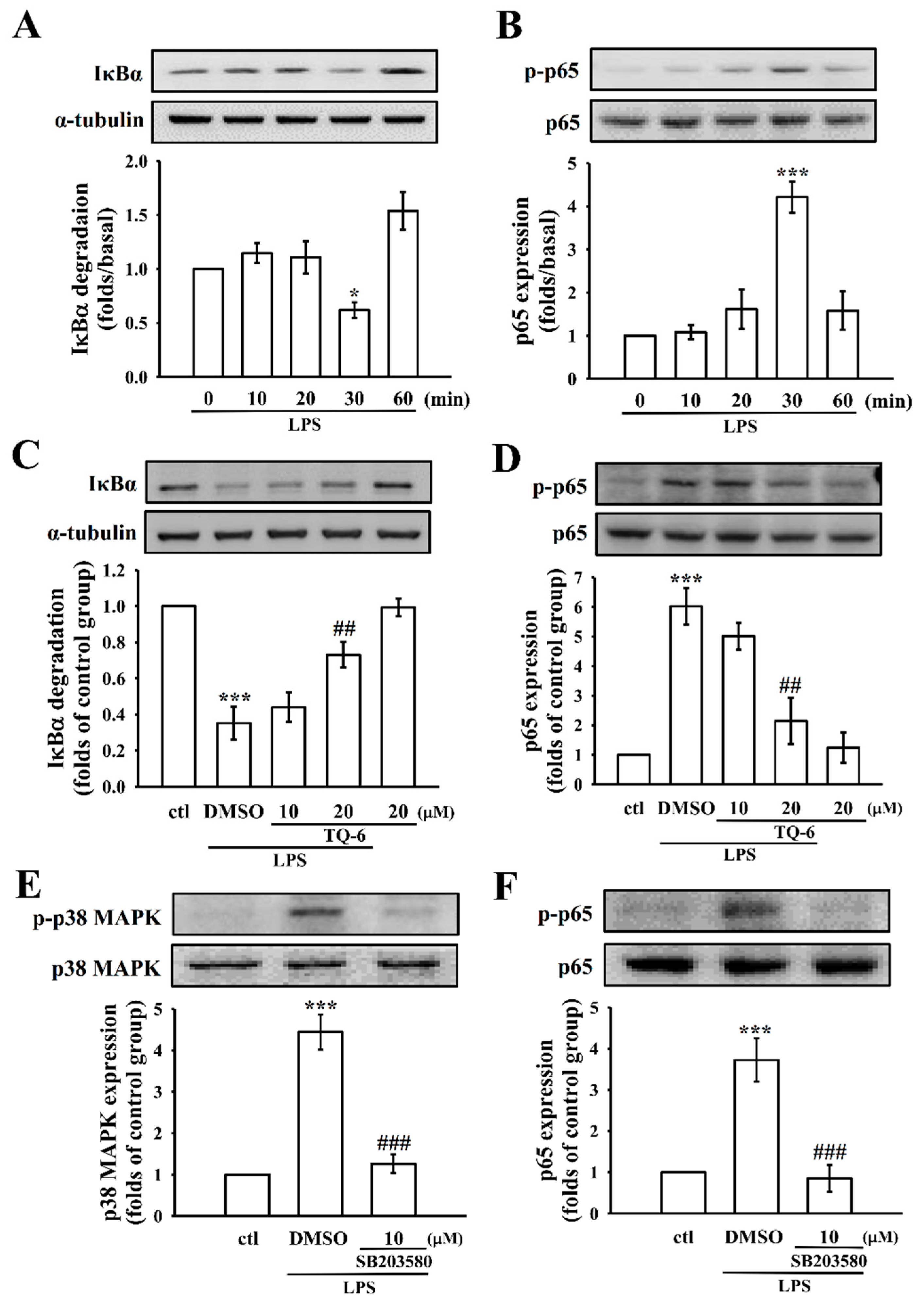
3.5. TQ-6 Regulates LPS-Induced NF-κB Activation in RAW Cells
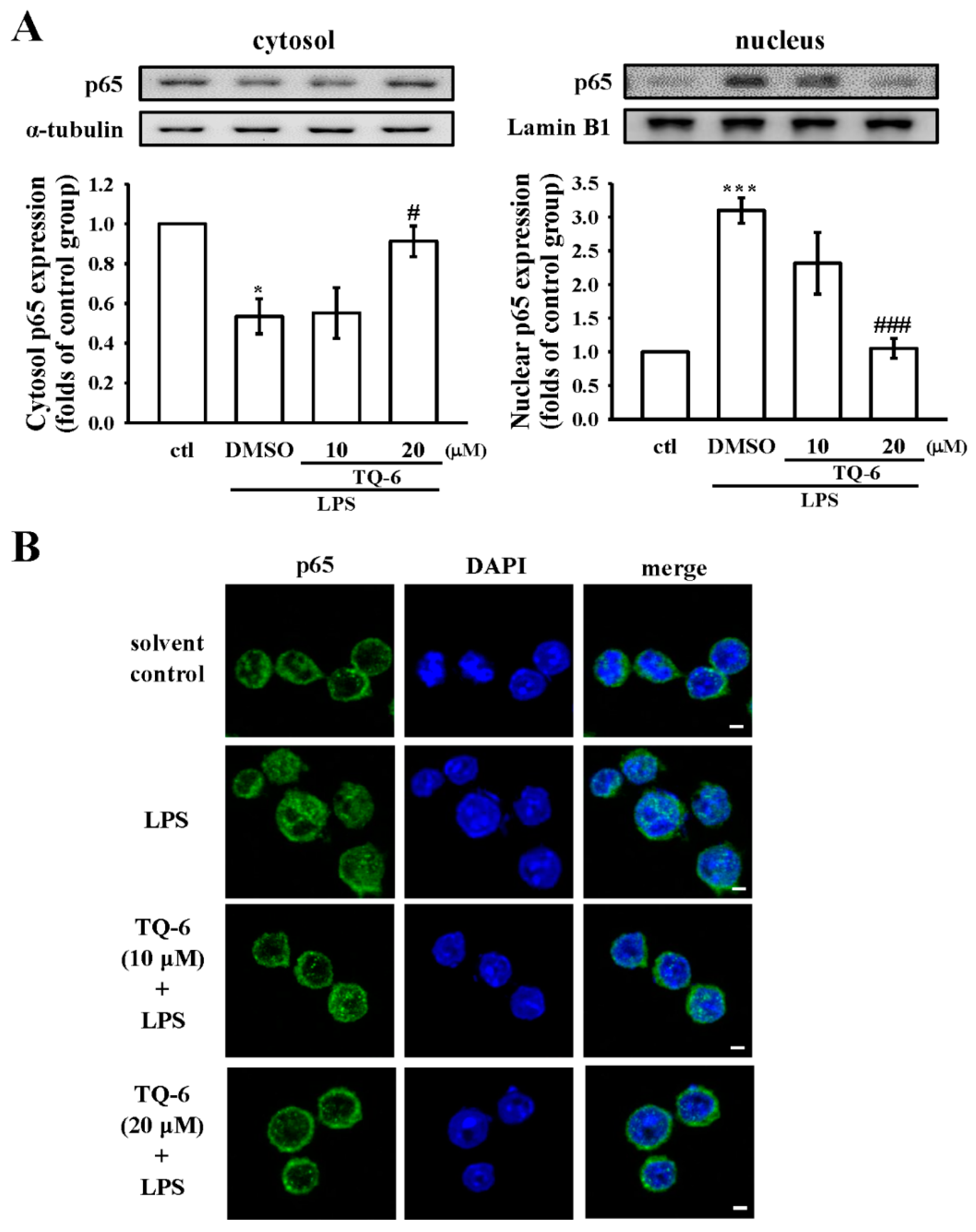
3.6. TQ-6 Attenuated, LPS-Induced NF-κB Nuclear Translocation in RAW Cells
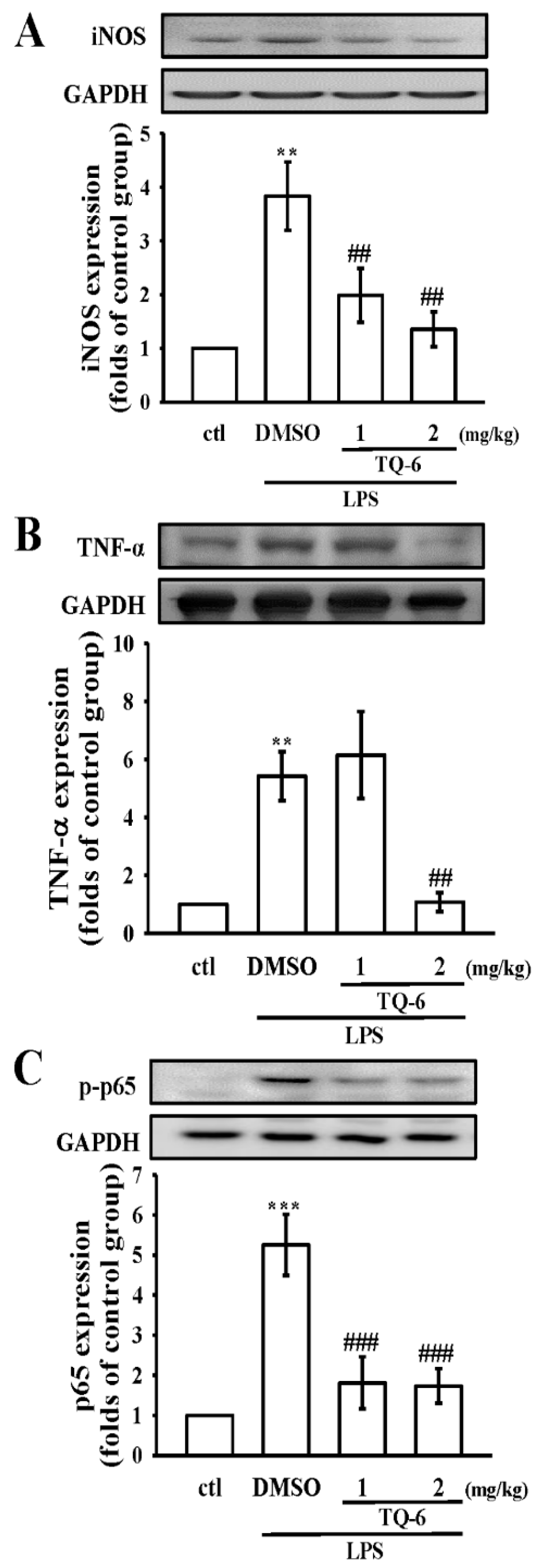
3.7. TQ-6 Restores Inflammation in LPS-Induced Liver Injury
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends. Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, M. Structural modifications of bacterial lipopolysaccharide that facilitate gramnegative bacteria evasion of host innate immunity. Front. Immunol. 2014, 4, 109. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, S. The role of the liver in sepsis. Int. Rev. Immunol. 2014, 33, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Anti-inflammatory agents: Present and future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Lin, S.; Zhong, H.J.; Ma, D.L. Metal complexes as potential modulators of inflammatory and autoimmune responses. Chem. Sci. 2015, 6, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Y.; Dong, H.; Zhang, C.Y.; Zhang, Y. Platinum-based agents for individualized cancer treatment. Curr. Mol. Med. 2013, 13, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Chohan, Z.H.; Iqbal, M.S.; Iqbal, H.S.; Scozzafava, A.; Supuran, C.T. Transition metal acetylsalicylates and their anti-inflammatory activity. J. Enzyme Inhib. Med. Chem. 2002, 17, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kale, M.A.; Shelke, R.; Nawale, R.B. Zinc-aceclofenac complex: Synthesis, hydrolysis study and antiinflammatory studies. Antiinflamm. Antiallergy Agents Med. Chem. 2014, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.H.; Velusamy, M.; Sheu, J.R.; Khamrang, T.; Jayakumar, T.; Lu, W.J.; Lin, K.H.; Chang, C.C. A novel ruthenium (II)-derived organometallic compound, TQ-6, potently inhibits platelet aggregation: Ex vivo and in vivo studies. Sci. Rep. 2017, 7, 9556. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Chen, Z.C.; Hsu, M.J.; Wang, S.H.; Jung, K.W.; Wu, W.F.; Pan, S.H.; Teng, R.D.; Yang, C.H.; Hsieh, C.Y. CME-1, a novel polysaccharide, suppresses iNOS expression in lipopolysaccharide-stimulated macrophages through ceramide-initiated protein phosphatase 2A activation. J. Cell. Mol. Med. 2018, 22, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.L.; Chen, R.J.; Jayakumar, T.; Lu, W.J.; Hsieh, C.Y.; Hsu, M.J.; Yang, C.H.; Chang, C.C.; Lin, Y.K.; Lin, K.H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Titheradge, M.A. Nitric oxide in septic shock. Biochim. Biophys. Acta 1999, 1411, 437–455. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.B.; McGahan, M.C.; Ferrell, J.B.; Adler, K.B.; Fleisher, L.N. Nitric oxide synthase inhibitors exert differential time-dependent effects on LPS-induced uveitis. Exp. Eye Res. 1996, 62, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Lin, S.; Chan, D.S.; Vong, C.T.; Hoi, P.M.; Wong, C.Y.; Ma, D.L.; Leung, C.H. A rhodium(III) complex inhibits LPS-induced nitric oxide production and angiogenic activity in cellulo. J. Inorg. Biochem. 2014, 140, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Möller, B.; Villiger, P.M. Inhibition of IL-1, IL-6, and TNF-alpha in immune-mediated inflammatory diseases. Springer Semin. Immunopathol. 2006, 27, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Cinel, I.; Opal, S.M. Molecular biology of inflammation and sepsis: A primer. Crit. Care Med. 2009, 37, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.J.; Wang, W.; Kang, T.S.; Yan, H.; Yang, Y.; Xu, L.; Wang, Y.; Ma, D.L.; Leung, C.H. A rhodium(III) complex as an inhibitor of neural precursor cell expressed, developmentally down-regulated 8-activating enzyme with in vivo activity against inflammatory bowel disease. J. Med. Chem. 2017, 60, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Serena, C.; Calvo, E.; Clares, M.P.; Diaz, M.L.; Chicote, J.U.; Beltrán-Debon, R.; Fontova, R.; Rodriguez, A.; García-España, E.; García-España, A. Significant in vivo anti-inflammatory activity of Pytren4Q-Mn a superoxide dismutase 2 (SOD2) mimetic scorpiand-like Mn (II) complex. PLoS ONE 2015, 10, e0119102. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, H.S.; Chong, Y.H.; Kang, J.L. p38 Mitogen-activated protein kinase up-regulates LPS-induced NF-kappaB activation in the development of lung injury and RAW 264.7 macrophages. Toxicology 2006, 225, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Satake, H.; Suzuki, K.; Aoki, T.; Otsuka, M.; Sugiura, Y.; Yamamoto, T.; Inoue, J. Cupric ion blocks NF-κB activation through inhibiting the signal induced phosphorylation of IκBα. Biochem. Biophys. Res. Commun. 1995, 216, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Jeong, J.Y.; Jue, D.M. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J. Immunol. 2000, 164, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Josephs, M.D.; Bahjat, F.R.; Fukuzuka, K.; Ksontini, R.; Solorzano, C.C.; Edwards, C.K., III; Tannahill, C.L.; MacKay, S.L.; Copeland, E.M., III; Moldawer, L.L. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1196–R1201. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Anderson, A.L.; Devereaux, M.W.; Fillon, S.A.; Harris, J.K.; Lovell, M.A.; Finegold, M.J.; Sokol, R.J. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 2012, 55, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Colletti, L.M.; Green, M. Lung and liver injury following hepatic ischemia/reperfusion in the rat is increased by exogenous lipopolysaccharide which also increases hepatic TNF production in vivo and in vitro. Shock 2001, 16, 312–319. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsia, C.-H.; Velusamy, M.; Jayakumar, T.; Chen, Y.-J.; Hsia, C.-W.; Tsai, J.-H.; Teng, R.-D.; Sheu, J.-R. Mechanisms of TQ-6, a Novel Ruthenium-Derivative Compound, against Lipopolysaccharide-Induced In Vitro Macrophage Activation and Liver Injury in Experimental Mice: The Crucial Role of p38 MAPK and NF-κB Signaling. Cells 2018, 7, 217. https://doi.org/10.3390/cells7110217
Hsia C-H, Velusamy M, Jayakumar T, Chen Y-J, Hsia C-W, Tsai J-H, Teng R-D, Sheu J-R. Mechanisms of TQ-6, a Novel Ruthenium-Derivative Compound, against Lipopolysaccharide-Induced In Vitro Macrophage Activation and Liver Injury in Experimental Mice: The Crucial Role of p38 MAPK and NF-κB Signaling. Cells. 2018; 7(11):217. https://doi.org/10.3390/cells7110217
Chicago/Turabian StyleHsia, Chih-Hsuan, Marappan Velusamy, Thanasekaran Jayakumar, Yen-Jen Chen, Chih-Wei Hsia, Jie-Heng Tsai, Ruei-Dun Teng, and Joen-Rong Sheu. 2018. "Mechanisms of TQ-6, a Novel Ruthenium-Derivative Compound, against Lipopolysaccharide-Induced In Vitro Macrophage Activation and Liver Injury in Experimental Mice: The Crucial Role of p38 MAPK and NF-κB Signaling" Cells 7, no. 11: 217. https://doi.org/10.3390/cells7110217
APA StyleHsia, C.-H., Velusamy, M., Jayakumar, T., Chen, Y.-J., Hsia, C.-W., Tsai, J.-H., Teng, R.-D., & Sheu, J.-R. (2018). Mechanisms of TQ-6, a Novel Ruthenium-Derivative Compound, against Lipopolysaccharide-Induced In Vitro Macrophage Activation and Liver Injury in Experimental Mice: The Crucial Role of p38 MAPK and NF-κB Signaling. Cells, 7(11), 217. https://doi.org/10.3390/cells7110217