Is there a Chance to Promote Arteriogenesis by DPP4 Inhibitors Even in Type 2 Diabetes? A Critical Review



Abstract
1. Introduction
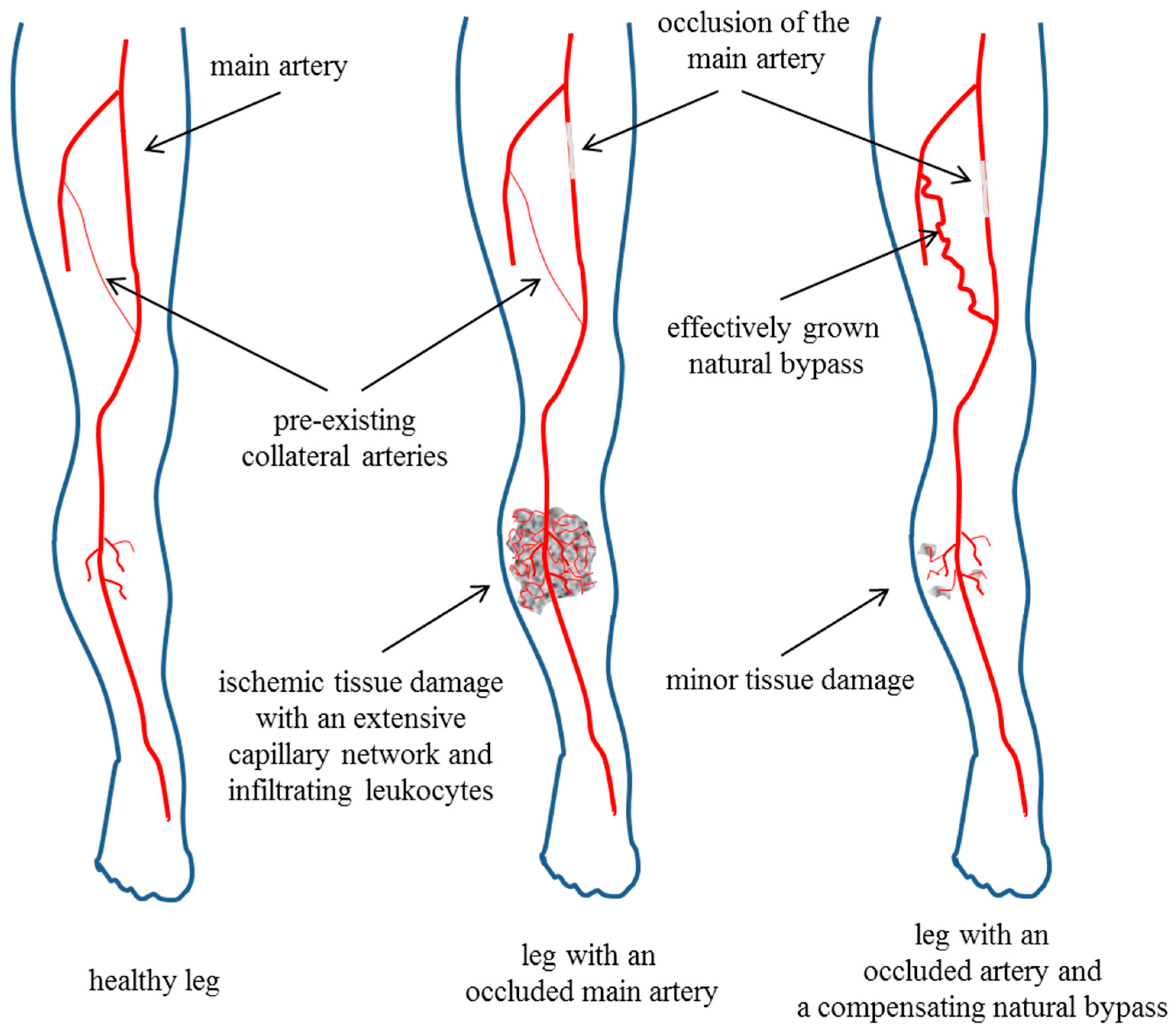
2. Arteriogenesis
3. Glucose Uptake and the Metabolic Disorder Diabetes Mellitus
3.1. Incretins, Insulin, and the Adrenergic System
3.2. Diabetes Mellitus and Glucose-Dependent Insulinotropic Peptide-1 (GLP-1)
4. Molecular Functions of Dipeptidyl-Peptidase-4 (DPP4)
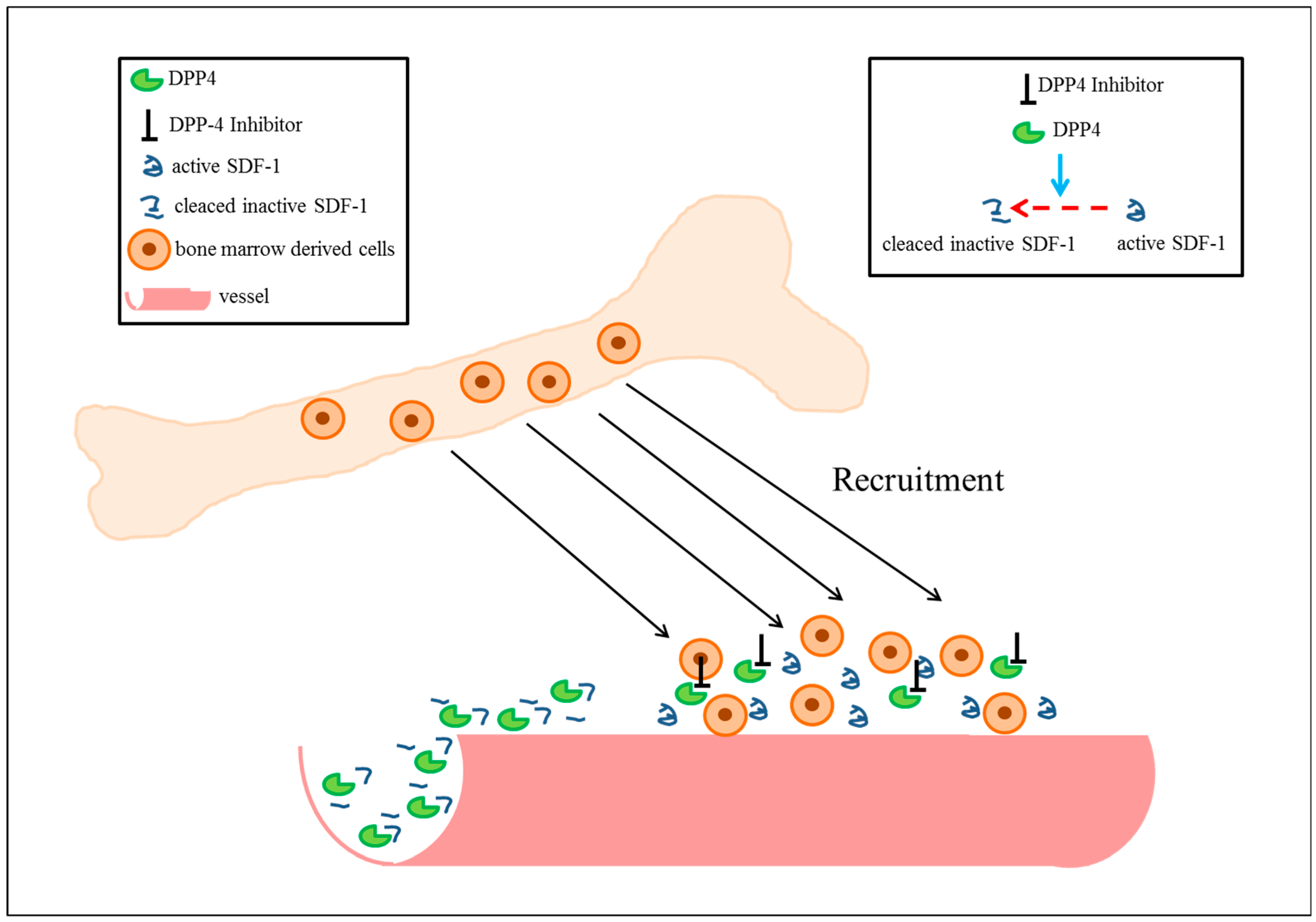
5. Cardiovascular Functions of Stromal-Cell-Derived Factor-1
6. DPP4 Inhibitors and Cardiovascular Diseases
6.1. DPP4 Inhibitors in Pre-Clinical Studies
6.2. DPP4 Inhibitors in Clinical Studies
6.3. DPP4 Inhibitors Revisited in Pre-Clinical Studies
6.4. Questions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World-Health-Organisation. Cardiovascular Diseases (cvds); World-Health-Organisation: Geneva, Switzerland, 2013. [Google Scholar]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Abaci, A.; Oguzhan, A.; Kahraman, S.; Eryol, N.K.; Unal, S.; Arinc, H.; Ergin, A. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation 1999, 99, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Rivard, A.; Silver, M.; Chen, D.; Kearney, M.; Magner, M.; Annex, B.; Peters, K.; Isner, J.M. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am. J. Pathol. 1999, 154, 355–363. [Google Scholar] [CrossRef]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biochem. Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Lasch, M.; Nekolla, K.; Klemm, A.H.; Buchheim, J.I.; Pohl, U.; Dietzel, S.; Deindl, E. Estimating hemodynamic shear stress in murine peripheral collateral arteries by two-photon line scanning. Mol. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Boehm, S.; Cai, W.J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.T.S.-R. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Florczyk, U.; Grochot-Przeczek, A.; Krist, B.; Loboda, A.; Jozkowicz, A.; Dulak, J. Limb ischemia and vessel regeneration: Is there a role for VEGF? Vasc. Pharmacol. 2016, 86, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Kofler, N.M.; Simons, M. Angiogenesis versus arteriogenesis: Neuropilin 1 modulation of VEGF signaling. F1000Prime Rep. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Nishio, M.; Peters, S.C.; Tschernatsch, M.; Walberer, M.; Weidemann, S.; Heidenreich, R.; Couraud, P.O.; Weksler, B.B.; Romero, I.A.; et al. Signaling mechanism of extracellular RNA in endothelial cells. FASEB J. 2009, 23, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Chandraratne, S.; von Bruehl, M.L.; Pagel, J.I.; Stark, K.; Kleinert, E.; Konrad, I.; Farschtschi, S.; Coletti, R.; Gartner, F.; Chillo, O.; et al. Critical role of platelet glycoprotein ibalpha in arterial remodeling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Ichikawa, N.; Lee, M.; Goto, M.; Sakai, H.; Kim, J.J.; Yoshida, M.; Handa, M.; Ikeda, Y.; Handa, S. Platelet surface P-selectin molecules increased after exposing platelet to a high shear flow. Int. Angiol. 2000, 19, 147–151. [Google Scholar] [PubMed]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Ziegelhoffer, T.; Kanse, S.M.; Fernandez, B.; Neubauer, E.; Carmeliet, P.; Preissner, K.T.; Schaper, W. Receptor-independent role of the urokinase-type plasminogen activator during arteriogenesis. FASEB J. 2003, 17, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Reichel, C.A.; Uhl, B.; Lerchenberger, M.; Puhr-Westerheide, D.; Rehberg, M.; Liebl, J.; Khandoga, A.; Schmalix, W.; Zahler, S.; Deindl, E.; et al. Urokinase-type plasminogen activator promotes paracellular transmigration of neutrophils via Mac-1, but independently of urokinase-type plasminogen activator receptor. Circulation 2011, 124, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Investig. 1998, 101, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Szade, A.; Grochot-Przeczek, A.; Florczyk, U.; Jozkowicz, A.; Dulak, J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life 2015, 67, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Goraya, T.Y.; Leibson, C.L.; Palumbo, P.J.; Weston, S.A.; Killian, J.M.; Pfeifer, E.A.; Jacobsen, S.J.; Frye, R.L.; Roger, V.L. Coronary atherosclerosis in diabetes mellitus: A population-based autopsy study. J. Am. Coll. Cardiol. 2002, 40, 946–953. [Google Scholar] [CrossRef]
- Weihrauch, D.; Lohr, N.L.; Mraovic, B.; Ludwig, L.M.; Chilian, W.M.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. Chronic hyperglycemia attenuates coronary collateral development and impairs proliferative properties of myocardial interstitial fluid by production of angiostatin. Circulation 2004, 109, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Peripheral Artery Disease in People with Diabetes. Diabetes Care 2003, 26, 3333–3341. [Google Scholar]
- Dolan, N.C.; Liu, K.; Criqui, M.H.; Greenland, P.; Guralnik, J.M.; Chan, C.; Schneider, J.R.; Mandapat, A.L.; Martin, G.; McDermott, M.M. Peripheral artery disease, diabetes, and reduced lower extremity functioning. Diabetes Care 2002, 25, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Prompers, L.; Schaper, N.; Apelqvist, J.; Edmonds, M.; Jude, E.; Mauricio, D.; Uccioli, L.; Urbancic, V.; Bakker, K.; Holstein, P.; et al. Prediction of outcome in individuals with diabetic foot ulcers: Focus on the differences between individuals with and without peripheral arterial disease. The EURODIALE Study. Diabetologia 2008, 51, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. (Lond.) 2010, 119, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Graham, K.; MacKenzie, E.J.; Ephraim, P.L.; Travison, T.G.; Brookmeyer, R. Estimating the prevalence of limb loss in the United States: 2005 to 2050. Arch. Phys. Med. Rehabil. 2008, 89, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Faglia, E.; Clerici, G.; Clerissi, J.; Gabrielli, L.; Losa, S.; Mantero, M.; Caminiti, M.; Curci, V.; Quarantiello, A.; Lupattelli, T.; et al. Long-term prognosis of diabetic patients with critical limb ischemia: A population-based cohort study. Diabetes Care 2009, 32, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Lombardi, A.; Sorriento, D.; Anastasio, A.; Del Giudice, C.; Formisano, P.; Beguinot, F.; Trimarco, B.; Miele, C.; Iaccarino, G. Age-related impairment in insulin release: The essential role of beta(2)-adrenergic receptor. Diabetes 2012, 61, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.H.; Jokubka, R.; Tojjar, D.; Granhall, C.; Hansson, O.; Li, D.Q.; Nagaraj, V.; Reinbothe, T.M.; Tuncel, J.; Eliasson, L.; et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 2010, 327, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, J.; Evans, B.A.; Bengtsson, T.; Summers, R.J. Multiple signalling pathways involved in beta2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br. J. Pharmacol. 2006, 147, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, D.S.; Bengtsson, T. alpha1A-adrenoceptors activate glucose uptake in L6 muscle cells through a phospholipase C-phosphatidylinositol-3 kinase-, and atypical protein kinase C.-dependent pathway. Endocrinology 2005, 146, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Lembo, G.; Capaldo, B.; Rendina, V.; Iaccarino, G.; Napoli, R.; Guida, R.; Trimarco, B.; Sacca, L. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am. J. Physiol. 1994, 266, E242–E247. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, E.; Del Giudice, C.; Santulli, G.; Trimarco, B.; Iaccarino, G. Opposite effects of beta2-adrenoceptor gene deletion on insulin signaling in liver and skeletal muscle. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Breda, E.; Oberg, A.L.; Powell, C.C.; Dalla Man, C.; Basu, A.; Vittone, J.L.; Klee, G.G.; Arora, P.; Jensen, M.D.; et al. Mechanisms of the age-associated deterioration in glucose tolerance: Contribution of alterations in insulin secretion, action, and clearance. Diabetes 2003, 52, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Beck-Nielsen, H.; Laakso, M.; Smith, U.; Yki-Jarvinen, H.; Ferrannini, E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J. Clin. Endocrinol. Metab. 1999, 84, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Santulli, G.; Campanile, A.; Galasso, G.; Cervero, P.; Altobelli, G.G.; Cimini, V.; Pastore, L.; Piscione, F.; Trimarco, B.; et al. Endothelial alpha1-adrenoceptors regulate neo-angiogenesis. Br. J. Pharmacol. 2008, 153, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Sorriento, D.; Cipolletta, E.; Santulli, G.; Fusco, A.; Zhou, R.H.; Eckhart, A.D.; Peppel, K.; Koch, W.J.; Trimarco, B.; et al. Impaired neoangiogenesis in beta(2)-adrenoceptor gene-deficient mice: Restoration by intravascular human beta(2)-adrenoceptor gene transfer and role of NFkappaB and CREB transcription factors. Br. J. Pharmacol. 2011, 162, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017; Centers for Disease Control and Prevention, US Department of Health and Human Services: Atlanta, GA, USA, 2017.
- Grant, P.J. Diabetes mellitus as a prothrombotic condition. J. Intern. Med. 2007, 262, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Blonde, L.; Klein, E.J.; Han, J.; Zhang, B.; Mac, S.M.; Poon, T.H.; Taylor, K.L.; Trautmann, M.E.; Kim, D.D.; Kendall, D.M. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes. Metab. 2006, 8, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Chehade, J.M.; Alcalde, R.; Naem, E.; Mooradian, A.D.; Wong, N.C.; Haas, M.J. Induction of apolipoprotein A-I gene expression by glucagon-like peptide-1 and exendin-4 in hepatocytes but not intestinal cells. Metabolism 2013, 62, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Irwin, N.; McClean, P.L.; Hunter, K.; Flatt, P.R. Metabolic effects of sustained activation of the GLP-1 receptor alone and in combination with background GIP receptor antagonism in high fat-fed mice. Diabetes Obes. Metab. 2009, 11, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Parlevliet, E.T.; Schroder-van der Elst, J.P.; Corssmit, E.P.; Picha, K.; O’Neil, K.; Stojanovic-Susulic, V.; Ort, T.; Havekes, L.M.; Romijn, J.A.; Pijl, H. CNTO736, a novel glucagon-like peptide-1 receptor agonist, ameliorates insulin resistance and inhibits very low-density lipoprotein production in high-fat-fed mice. J. Pharmacol. Exp. Ther. 2009, 328, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Shen, H.; Liu, M.; Yang, Q.; Zheng, S.; Sabo, M.; D’Alessio, D.A.; Tso, P. GLP-1 reduces intestinal lymph flow, triglyceride absorption, and apolipoprotein production in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G943–G949. [Google Scholar] [CrossRef] [PubMed]
- Vanderweele, D.A.; Geiselman, P.J.; Novin, D. Pancreatic glucagon, food deprivation and feeding in intact and vagotomized rabbits. Physiol. Behav. 1979, 23, 155–158. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Kirino, Y.; Sei, M.; Kawazoe, K.; Minakuchi, K.; Sato, Y. Plasma dipeptidyl peptidase 4 activity correlates with body mass index and the plasma adiponectin concentration in healthy young people. Endocr. J. 2012, 59, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Lamers, D.; Famulla, S.; Wronkowitz, N.; Hartwig, S.; Lehr, S.; Ouwens, D.M.; Eckardt, K.; Kaufman, J.M.; Ryden, M.; Muller, S.; et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011, 60, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Bullock, B.P.; Heller, R.S.; Habener, J.F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 1996, 137, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, D.A.; Kahn, S.E.; Leusner, C.R.; Ensinck, J.W. Glucagon-like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J. Clin. Investig. 1994, 93, 2263–2266. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, M.; Rebelos, E.; Gastaldelli, A.; Astiarraga, B.D.; Casolaro, A.; Barsotti, E.; Pocai, A.; Nauck, M.; Muscelli, E.; Ferrannini, E. Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia 2013, 56, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Iltz, J.L.; Baker, D.E.; Setter, S.M.; Keith Campbell, R. Exenatide: An incretin mimetic for the treatment of type 2 diabetes mellitus. Clin. Ther. 2006, 28, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar] [PubMed]
- Dhanesha, N.; Joharapurkar, A.; Shah, G.; Dhote, V.; Kshirsagar, S.; Bahekar, R.; Jain, M. Exendin-4 reduces glycemia by increasing liver glucokinase activity: An insulin independent effect. Pharmacol. Rep. 2012, 64, 140–149. [Google Scholar] [CrossRef]
- Kolterman, O.G.; Buse, J.B.; Fineman, M.S.; Gaines, E.; Heintz, S.; Bicsak, T.A.; Taylor, K.; Kim, D.; Aisporna, M.; Wang, Y.; et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Degn, K.B.; Brock, B.; Juhl, C.B.; Djurhuus, C.B.; Grubert, J.; Kim, D.; Han, J.; Taylor, K.; Fineman, M.; Schmitz, O. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes 2004, 53, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Gedulin, B.R.; Nikoulina, S.E.; Smith, P.A.; Gedulin, G.; Nielsen, L.L.; Baron, A.D.; Parkes, D.G.; Young, A.A. Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology 2005, 146, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Huang, T.; Chen, A.; Chen, X.; Wang, L.; Shen, F.; Gu, X. Glucagon-like peptide 1 improves insulin resistance in vitro through anti-inflammation of macrophages. Braz. J. Med. Biol. Res. 2016, 49, e5826. [Google Scholar] [CrossRef] [PubMed]
- Idris, I.; Patiag, D.; Gray, S.; Donnelly, R. Exendin-4 increases insulin sensitivity via a PI-3-kinase-dependent mechanism: Contrasting effects of GLP-1. Biochem. Pharmacol. 2002, 63, 993–996. [Google Scholar] [CrossRef]
- Wang, A.; Li, T.; An, P.; Yan, W.; Zheng, H.; Wang, B.; Mu, Y. Exendin-4 Upregulates Adiponectin Level in Adipocytes via Sirt1/Foxo-1 Signaling Pathway. PLoS ONE 2017, 12, e0169469. [Google Scholar] [CrossRef] [PubMed]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Daiber, A.; Oelze, M.; Brandt, M.; Closs, E.; Xu, J.; Thum, T.; Bauersachs, J.; Ertl, G.; Zou, M.H.; et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis 2008, 198, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Shah, Z.; Pineda, C.; Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Racoma, I.; Deiuliis, J.; Xu, X.; Sun, Q.; Moffatt-Bruce, S.; et al. Acute DPP-4 inhibition modulates vascular tone through GLP-1 independent pathways. Vasc. Pharmacol. 2011, 55, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.Y.; Riviere, P.J.; Trojnar, J.; Junien, J.L.; Akinsanya, K.O. Cloning and characterization of dipeptidyl peptidase 10, a new member of an emerging subgroup of serine proteases. Biochem. J. 2003, 373, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Polgar, L. The prolyl oligopeptidase family. Cell. Mol. Life Sci. 2002, 59, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Polgar, L.; Barrett, A.J. A new family of serine-type peptidases related to prolyl oligopeptidase. Biochem. J. 1991, 279 Pt 3, 907–908. [Google Scholar] [CrossRef]
- Sedo, A.; Malik, R. Dipeptidyl peptidase IV-like molecules: Homologous proteins or homologous activities? Biochim. Biophys. Acta 2001, 1550, 107–116. [Google Scholar] [CrossRef]
- Niedermeyer, J.; Enenkel, B.; Park, J.E.; Lenter, M.; Rettig, W.J.; Damm, K.; Schnapp, A. Mouse fibroblast-activation protein—Conserved Fap gene organization and biochemical function as a serine protease. Eur. J. Biochem. 1998, 254, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Jacotot, E.; Callebaut, C.; Blanco, J.; Krust, B.; Neubert, K.; Barth, A.; Hovanessian, A.G. Dipeptidyl-peptidase IV-beta, a novel form of cell-surface-expressed protein with dipeptidyl-peptidase IV activity. Eur. J. Biochem. 1996, 239, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Yokotani, N.; Hunter, C.; Doi, K.; Wenthold, R.J.; Shimasaki, S. Differential expression of two distinct forms of mRNA encoding members of a dipeptidyl aminopeptidase family. Proc. Natl. Acad. Sci. USA 1992, 89, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.M.; Fukasawa, K.; Higaki, K.; Shiina, N.; Ohno, M.; Ito, S.; Otogoto, J.; Ota, N. Cloning and functional expression of rat kidney dipeptidyl peptidase II. Biochem. J. 2001, 353, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.A.; Yu, D.M.; Woollatt, E.; Sutherland, G.R.; McCaughan, G.W.; Gorrell, M.D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 2000, 267, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Grigo, C.; Steinbeck, J.; von Horsten, S.; Amann, K.; Daniel, C. Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides 2014, 57, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R. Dipeptidyl-peptidase IV (CD26)—Role in the inactivation of regulatory peptides. Regul. Pept. 1999, 85, 9–24. [Google Scholar] [CrossRef]
- Zhong, J.; Rao, X.; Deiuliis, J.; Braunstein, Z.; Narula, V.; Hazey, J.; Mikami, D.; Needleman, B.; Satoskar, A.R.; Rajagopalan, S. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes 2013, 62, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, C.; Schlossman, S.F. The structure and function of CD26 in the T-cell immune response. Immunol. Rev. 1998, 161, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, R.; Woods Ignatoski, K.M.; Okawada, M.; Hartmann, B.; Holst, J.; Teitelbaum, D.H. Stimulation of intestinal growth and function with DPP4 inhibition in a mouse short bowel syndrome model. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G410–G419. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Arredondo-Vega, F.X.; Santisteban, I.; Kelly, S.J.; Patel, D.D.; Hershfield, M.S. The binding site of human adenosine deaminase for CD26/Dipeptidyl peptidase IV: The Arg142Gln mutation impairs binding to cd26 but does not cause immune deficiency. J. Exp. Med. 2000, 192, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Schwartz, F.; Entschladen, F.; Niggemann, B.; Zaenker, K.S. DPPIV inhibitors extend GLP-2 mediated tumour promoting effects on intestinal cancer cells. Regul. Pept. 2006, 137, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B.; Hughes, T.E. Inhibition of dipeptidyl peptidase-4 augments insulin secretion in response to exogenously administered glucagon-like peptide-1, glucose-dependent insulinotropic polypeptide, pituitary adenylate cyclase-activating polypeptide, and gastrin-releasing peptide in mice. Endocrinology 2005, 146, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Devin, J.K.; Pretorius, M.; Nian, H.; Yu, C.; Billings FTt and Brown, N.J. Dipeptidyl-peptidase 4 inhibition and the vascular effects of glucagon-like peptide-1 and brain natriuretic peptide in the human forearm. J. Am. Heart. Assoc. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Stremenova, J.; Krepela, E.; Sedo, A. Modulation of substance P signaling by dipeptidyl peptidase-IV enzymatic activity in human glioma cell lines. Physiol. Res. 2008, 57, 443–449. [Google Scholar] [PubMed]
- Reinehr, T.; Roth, C.L.; Enriori, P.J.; Masur, K. Changes of dipeptidyl peptidase IV (DPP-IV) in obese children with weight loss: Relationships to peptide, Y.Y.; pancreatic peptide, and insulin sensitivity. J. Pediatr. Endocrinol. Metab. 2010, 23, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.; Kaestner, F.; Wolf, R.; Stiller, H.; Heiser, U.; Manhart, S.; Hoffmann, T.; Rahfeld, J.U.; Demuth, H.U.; Rothermundt, M.; et al. Identifying neuropeptide Y (NPY) as the main stress-related substrate of dipeptidyl peptidase 4 (DPP4) in blood circulation. Neuropeptides 2016, 57, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Ajami, K.; Pitman, M.R.; Wilson, C.H.; Park, J.; Menz, R.I.; Starr, A.E.; Cox, J.H.; Abbott, C.A.; Overall, C.M.; Gorrell, M.D. Stromal cell-derived factors 1alpha and 1beta, inflammatory protein-10 and interferon-inducible T cell chemo-attractant are novel substrates of dipeptidyl peptidase 8. FEBS Lett. 2008, 582, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Fontarigo, J.J.; Gonzalez-Barcala, F.J.; San-Jose, M.E.; Cruz, M.J.; Linares, T.; Soto-Mera, M.T.; Valdes-Cuadrado, L.; Garcia-Gonzalez, M.A.; Andrade-Bulos, L.J.; Arias, P.; et al. Expansion of a CD26low Effector TH Subset and Reduction in Circulating Levels of sCD26 in Stable Allergic Asthma in Adults. J. Investig. Allergol. Clin. Immunol. 2018, 28, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Schutyser, E.; Menten, P.; Struyf, S.; Wuyts, A.; Opdenakker, G.; Detheux, M.; Parmentier, M.; Durinx, C.; Lambeir, A.M.; et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 2001, 98, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Yamaguchi, N.; Munakata, Y.; Ikushima, H.; Lee, J.F.; Hosono, O.; Schlossman, S.F.; Morimoto, C. CD26/dipeptidyl peptidase IV differentially regulates the chemotaxis of T cells and monocytes toward RANTES: Possible mechanism for the switch from innate to acquired immune response. Int. Immunol. 1999, 11, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Hoggatt, J.; O’Leary, H.A.; Mantel, C.; Chitteti, B.R.; Cooper, S.; Messina-Graham, S.; Hangoc, G.; Farag, S.; Rohrabaugh, S.L.; et al. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat. Med. 2012, 18, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, R.; Martinez-Navio, J.M.; Lejeune, M.; Climent, N.; Oliva, H.; Gatell, J.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 9583–9588. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Uchiyama, M.; Yamochi, T.; Nishibashi, K.; Hosono, O.; Takahashi, N.; Kina, S.; Tanaka, H.; Lin, X.; Dang, N.H.; et al. Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J. Biol. Chem. 2007, 282, 10117–10131. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Munakata, Y.; Ishii, T.; Iwata, S.; Terashima, M.; Tanaka, H.; Schlossman, S.F.; Morimoto, C. Internalization of CD26 by mannose 6-phosphate/insulin-like growth factor II receptor contributes to T. cell activation. Proc. Natl. Acad. Sci. USA 2000, 97, 8439–8444. [Google Scholar] [CrossRef] [PubMed]
- Salgado, F.J.; Lojo, J.; Alonso-Lebrero, J.L.; Lluis, C.; Franco, R.; Cordero, O.J.; Nogueira, M. A role for interleukin-12 in the regulation of T cell plasma membrane compartmentation. J. Biol. Chem. 2003, 278, 24849–24857. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, G.; Zhao, Q.; Salamone, M.; Yeh, Y.; Zucker, S.; Chen, W.T. The protease complex consisting of dipeptidyl peptidase, I.V.; seprase plays a role in the migration and invasion of human endothelial cells in collagenous matrices. Cancer Res. 2006, 66, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Yamochi, T.; Uchiyama, M.; Nishibashi, K.; Yoshikawa, N.; Shimizu, N.; Iwata, S.; Tanaka, H.; Dang, N.H.; Morimoto, C. CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc. Natl. Acad. Sci. USA 2004, 101, 14186–14191. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Degray, B.C.; Nagy, T.; Biemesderfer, D.; Aronson, P.S. Association of Na(+)-H(+) exchanger isoform NHE3 and dipeptidyl peptidase IV in the renal proximal tubule. J. Biol. Chem. 2001, 276, 46671–46677. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, W.A.; Liu, J.; Reutter, W.; Saenger, W.; Fan, H. Crystal structures of HIV-1 Tat-derived nonapeptides Tat-(1-9) and Trp2-Tat-(1-9) bound to the active site of dipeptidyl-peptidase IV (CD26). J. Biol. Chem. 2005, 280, 14911–14917. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; DiCorleto, P.E.; et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Deglurkar, I.; Mal, N.; Mills, W.R.; Popovic, Z.B.; McCarthy, P.; Blackstone, E.H.; Laurita, K.R.; Penn, M.S. Mechanical and electrical effects of cell-based gene therapy for ischemic cardiomyopathy are independent. Hum. Gene Ther. 2006, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, S.; Miller, T.J.; Pastore, J.M.; Kiedrowski, M.; Aras, R.; Penn, M.S. Plasmid-based transient human stromal cell-derived factor-1 gene transfer improves cardiac function in chronic heart failure. Gene Ther. 2011, 18, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mal, N.; Kiedrowski, M.; Chacko, M.; Askari, A.T.; Popovic, Z.B.; Koc, O.N.; Penn, M.S. SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. FASEB J. 2007, 21, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.W.; Hangoc, G.; Mantel, C.R.; Broxmeyer, H.E. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science 2004, 305, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Zaruba, M.M.; Theiss, H.D.; Vallaster, M.; Mehl, U.; Brunner, S.; David, R.; Fischer, R.; Krieg, L.; Hirsch, E.; Huber, B.; et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell 2009, 4, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Dingenouts, C.K.E.; Bakker, W.; Lodder, K.; Wiesmeijer, K.C.; Moerkamp, A.T.; Maring, J.A.; Arthur, H.M.; Smits, A.M.; Goumans, M.J. Inhibiting DPP4 in a mouse model of HHT1 results in a shift towards regenerative macrophages and reduces fibrosis after myocardial infarction. PLoS ONE 2017, 12, e0189805. [Google Scholar] [CrossRef] [PubMed]
- Haverslag, R.T.; de Groot, D.; Grundmann, S.; Meder, B.; Goumans, M.J.; Pasterkamp, G.; Hoefer, I.E.; de Kleijn, D.P. CD26 inhibition enhances perfusion recovery in ApoE-/-mice. Curr. Vasc. Pharmacol. 2013, 11, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Kollet, O.; Ponomaryov, T.; Petit, I.; Franitza, S.; Grabovsky, V.; Slav, M.M.; Nagler, A.; Lider, O.; Alon, R.; et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: Role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 2000, 95, 3289–3296. [Google Scholar] [PubMed]
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 1998, 273, 23169–23175. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.N.; Kalnins, A.; Andrassy, M.; Wagner, A.; Klussmann, S.; Rentsch, M.; Habicht, A.; Pratschke, S.; Stangl, M.; Bazhin, A.V.; et al. SDF-1/CXCR4/CXCR7 is pivotal for vascular smooth muscle cell proliferation and chronic allograft vasculopathy. Transpl. Int. 2015, 28, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- De Groot, D.; Haverslag, R.T.; Pasterkamp, G.; de Kleijn, D.P.; Hoefer, I.E. Targeted deletion of the inhibitory NF-kappaB p50 subunit in bone marrow-derived cells improves collateral growth after arterial occlusion. Cardiovasc. Res. 2010, 88, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Eitenmuller, I.; Volger, O.; Kluge, A.; Troidl, K.; Barancik, M.; Cai, W.J.; Heil, M.; Pipp, F.; Fischer, S.; Horrevoets, A.J.; et al. The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 2006, 99, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Bray, E.J.; Abouhamze, Z.; Moldawer, L.L.; Voskuil, M.; Piek, J.J.; Buschmann, I.R.; Ozaki, C.K. Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation 2002, 105, 1639–1641. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, N.; Hoefer, I.; Buschmann, I.; Heil, M.; Kostin, S.; Deindl, E.; Vogel, S.; Korff, T.; Augustin, H.; Bode, C.; et al. Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J. 2002. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.R.; Ogle, M.E.; McFaline-Figueroa, J.; Segar, C.E.; Temenoff, J.S.; Botchwey, E.A. Spatially localized recruitment of anti-inflammatory monocytes by SDF-1alpha-releasing hydrogels enhances microvascular network remodeling. Biomaterials 2015, 77, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Troidl, C.; Jung, G.; Troidl, K.; Hoffmann, J.; Mollmann, H.; Nef, H.; Schaper, W.; Hamm, C.W.; Schmitz-Rixen, T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr. Vasc. Pharmacol. 2013, 11, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Franz, W.M.; Kuhlenthal, S.; Kuschnerus, K.; Remm, F.; Gross, L.; Theiss, H.D.; Landmesser, U.; Krankel, N. DPP-4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int. J. Cardiol. 2015, 199, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Shin, S.; Shigihara, T.; Hahm, E.; Liu, M.J.; Han, J.; Yoon, J.W.; Jun, H.S. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007, 56, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Van Weel, V.; de Vries, M.; Voshol, P.J.; Verloop, R.E.; Eilers, P.H.; van Hinsbergh, V.W.; van Bockel, J.H.; Quax, P.H. Hypercholesterolemia reduces collateral artery growth more dominantly than hyperglycemia or insulin resistance in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, I.R.; Hoefer, I.E.; van Royen, N.; Katzer, E.; Braun-Dulleaus, R.; Heil, M.; Kostin, S.; Bode, C.; Schaper, W. GM-CSF: A strong arteriogenic factor acting by amplification of monocyte function. Atherosclerosis 2001, 159, 343–356. [Google Scholar] [CrossRef]
- Deindl, E.; Zaruba, M.M.; Brunner, S.; Huber, B.; Mehl, U.; Assmann, G.; Hoefer, I.E.; Mueller-Hoecker, J.; Franz, W.M. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006, 20, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Hemingway, H.; Lansky, A.J.; Knapp, G.; Pitt, B.; Seiler, C. The impact of the coronary collateral circulation on mortality: A meta-analysis. Eur. Heart. J. 2012, 33, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Fazel, S.; Cimini, M.; Chen, L.; Li, S.; Angoulvant, D.; Fedak, P.; Verma, S.; Weisel, R.D.; Keating, A.; Li, R.K. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J. Clin. Investig. 2006, 116, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Boucaud-Maitre, D. Inclusion of the EXAMINE study in a meta-analysis of the addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia. BMJ 2016, 353, i3186. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Cardiovascular Effects of New Oral Glucose-Lowering Agents: DPP-4 and SGLT-2 Inhibitors. Circ. Res. 2018, 122, 1439–1459. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Marx, N.; Kahn, S.E.; Zinman, B.; Kastelein, J.J.; Lachin, J.M.; Bluhmki, E.; Patel, S.; Johansen, O.E.; Woerle, H.J. Cardiovascular outcome trials in type 2 diabetes and the sulphonylurea controversy: Rationale for the active-comparator CAROLINA trial. Diabetes Vasc. Dis. Res. 2013, 10, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Jawa, A.; Kcomt, J.; Fonseca, V.A. Diabetic nephropathy and retinopathy. Med. Clin. North Am. 2004, 88, 1001–1036. [Google Scholar] [CrossRef] [PubMed]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Wang, J.; Wu, H.Y.; Chien, K.L.; Hung, K.Y. Dipeptidyl peptidase 4 inhibitor use is associated with a lower risk of incident acute kidney injury in patients with diabetes. Oncotarget 2017, 8, 53028–53040. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.R.; Park, S.W.; Kim, J.W.; Kim, J.H.; Lee, K. Protective Effects of Dipeptidyl Peptidase-4 Inhibitors on Progression of Diabetic Retinopathy in Patients with Type 2 Diabetes. Retina 2016, 36, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Almeida, L.; Silva, A.P.; Fontes-Ribeiro, C.; Ambrosio, A.F.; Cristovao, A.; Fernandes, R. The dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin ameliorates retinal endothelial cell dysfunction triggered by inflammation. Biomed. Pharmacother. 2018, 102, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Kanozawa, K.; Noguchi, Y.; Sugahara, S.; Nakamura, S.; Yamamoto, H.; Kaneko, K.; Kono, R.; Sato, S.; Ogawa, T.; Hasegawa, H.; et al. The renoprotective effect and safety of a DPP-4 inhibitor, sitagliptin, at a small dose in type 2 diabetic patients with a renal dysfunction when changed from other DPP-4 inhibitors: REAL trial. Clin. Exp. Nephrol. 2018, 22, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.S. An angiotensin-converting enzyme inhibitor modulates stromal-derived factor-1 through CD26/dipeptidyl peptidase IV to inhibit laser-induced choroidal neovascularization. Mol. Vis. 2013, 19, 1107–1121. [Google Scholar] [PubMed]
- Theiss, H.D.; Gross, L.; Vallaster, M.; David, R.; Brunner, S.; Brenner, C.; Nathan, P.; Assmann, G.; Mueller-Hoecker, J.; Vogeser, M.; et al. Antidiabetic gliptins in combination with G-CSF enhances myocardial function and survival after acute myocardial infarction. Int. J. Cardiol. 2013, 168, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Remm, F.; Krankel, N.; Lener, D.; Drucker, D.J.; Sopper, S.; Brenner, C. Sitagliptin Accelerates Endothelial Regeneration after Vascular Injury Independent from GLP1 Receptor Signaling. Stem Cells Int. 2018, 2018, 5284963. [Google Scholar] [CrossRef] [PubMed]
- Moraes, F.; Paye, J.; Mac Gabhann, F.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Bluher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018, 555, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Aschner, P.; Kipnes, M.S.; Lunceford, J.K.; Sanchez, M.; Mickel, C.; Williams-Herman, D.E. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006, 29, 2632–2637. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Park, C.Y.; Ahn, K.J.; Kim, N.H.; Jang, H.C.; Lee, M.K.; Park, J.Y.; Chung, C.H.; Min, K.W.; Sung, Y.A.; et al. A randomized, double-blind, placebo-controlled, phase II clinical trial to investigate the efficacy and safety of oral DA-1229 in patients with type 2 diabetes mellitus who have inadequate glycaemic control with diet and exercise. Diabetes Metab. Res. Rev. 2015, 31, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Kondo, K. Efficacy, safety and dose-response relationship of teneligliptin, a dipeptidyl peptidase-4 inhibitor, in Japanese patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2013, 15, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Kutoh, E.; Ukai, Y. Alogliptin as an initial therapy in patients with newly diagnosed, drug naive type 2 diabetes: A randomized, control trial. Endocrine 2012, 41, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Raz, I.; Hanefeld, M.; Xu, L.; Caria, C.; Williams-Herman, D.; Khatami, H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006, 49, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Ristic, S.; Byiers, S.; Foley, J.; Holmes, D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: Vildagliptin (LAF237) dose response. Diabetes Obes. Metab. 2005, 7, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y.; McCormack, P.L. Exenatide Extended-Release: An Updated Review of Its Use in Type 2 Diabetes Mellitus. Drugs 2015, 75, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Cao, X.; Zhou, M.; Zou, P.; Hu, J. Efficacy and safety of once-weekly semaglutide for the treatment of type 2 diabetes. Expert Opin. Investig. Drugs 2017, 26, 1083–1089. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Function | Protein | Result of DPP4 Cleavage |
|---|---|---|
| Regulatory peptides | GLP-1 | Inactivation |
| GLP-2 | Inactivation | |
| GIP | Inactivation | |
| GRP | Inactivation | |
| GHRF | Inactivation | |
| Neuropeptides | BNP | Activity reduced |
| Substance P | Inactivation | |
| Peptide YY | Receptor specificity | |
| NPY | Receptor specificity | |
| Chemokines | ITAC | Inactivation |
| IP-10 | Inactivation | |
| Eotaxin | Inactivation | |
| MIG | Altered cell type specificity | |
| MDC | Inactivation | |
| RANTES | Inactivation | |
| G-CSF | Inactivation | |
| GM-CSF | Inactivation |
| Study | Endpoint |
|---|---|
| SAVOR-TIMI, Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Type 2 Diabetes Mellitus [133] | cardiovascular death, nonfatal myocardial infarction, nonfatal stroke |
| TECOS, Trial Evaluating Cardiovascular Outcomes with Sitagliptin [132] | cardiovascular death, nonfatal myocardial infarction, nonfatal stroke |
| EXAMINE, Examination of Cardiovascular Outcomes: Alogliptin vs. Standard Care in Patients with Type 2 Diabetes Mellitus and Acute Coronary Syndrome [134] | cardiovascular death, nonfatal myocardial infarction, nonfatal stroke |
| CAROLINA, Cardiovascular Outcome Study of Linagliptin v.s. Glimepride in Patients with Type 2 Diabetes Mellitus [136] | cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, unstable angina pectoris |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vedantham, S.; Kluever, A.-K.; Deindl, E. Is there a Chance to Promote Arteriogenesis by DPP4 Inhibitors Even in Type 2 Diabetes? A Critical Review. Cells 2018, 7, 181. https://doi.org/10.3390/cells7100181
Vedantham S, Kluever A-K, Deindl E. Is there a Chance to Promote Arteriogenesis by DPP4 Inhibitors Even in Type 2 Diabetes? A Critical Review. Cells. 2018; 7(10):181. https://doi.org/10.3390/cells7100181
Chicago/Turabian StyleVedantham, Srinivasan, Anna-Kristina Kluever, and Elisabeth Deindl. 2018. "Is there a Chance to Promote Arteriogenesis by DPP4 Inhibitors Even in Type 2 Diabetes? A Critical Review" Cells 7, no. 10: 181. https://doi.org/10.3390/cells7100181
APA StyleVedantham, S., Kluever, A.-K., & Deindl, E. (2018). Is there a Chance to Promote Arteriogenesis by DPP4 Inhibitors Even in Type 2 Diabetes? A Critical Review. Cells, 7(10), 181. https://doi.org/10.3390/cells7100181