Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma

 , , , ,
, , , , 
 ,
,
Abstract
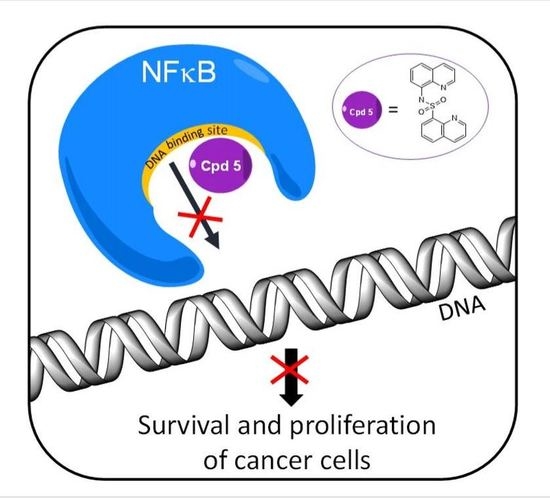
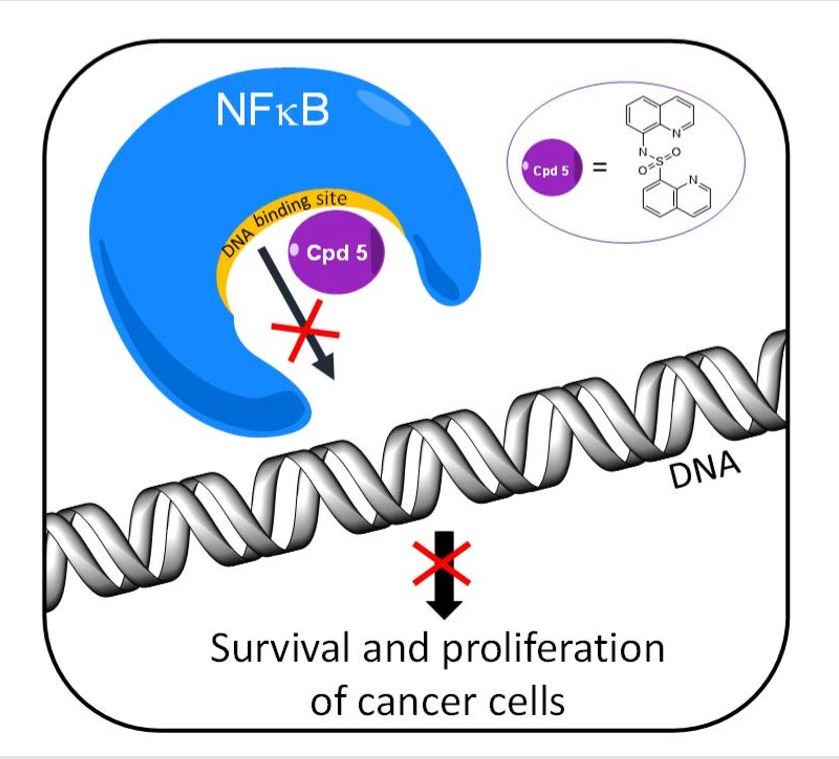
1. Introduction
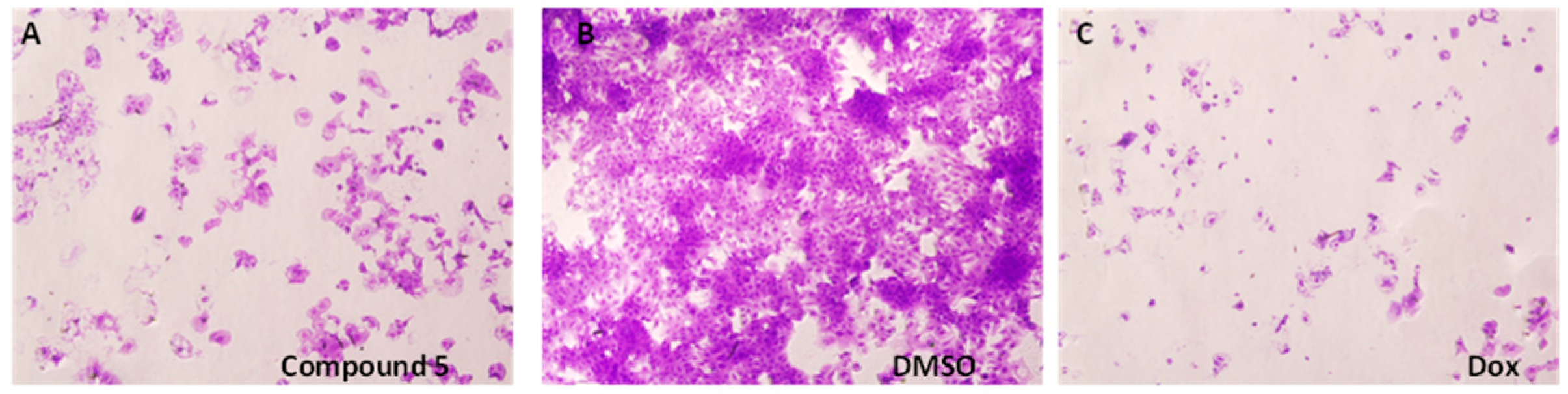
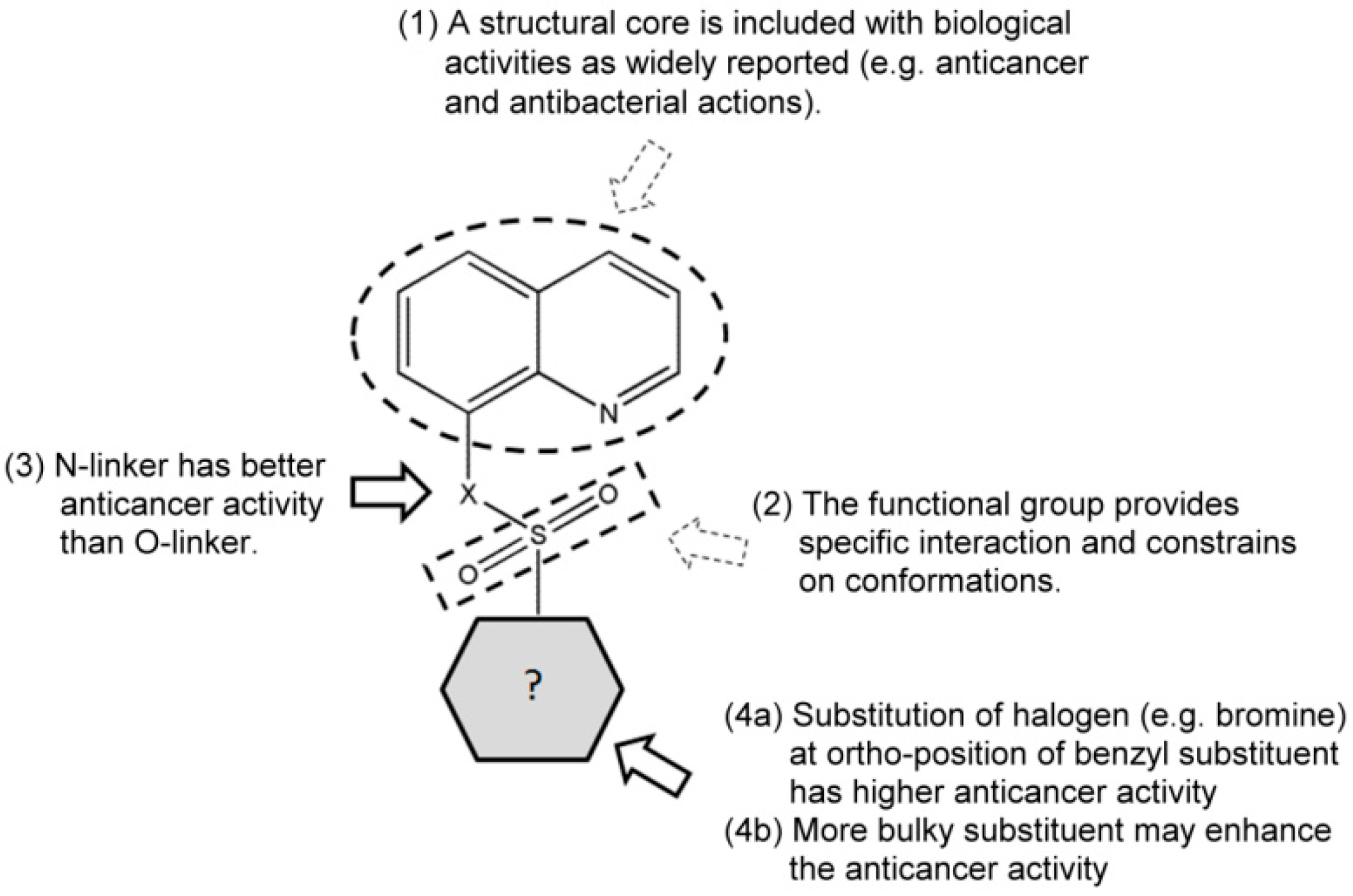
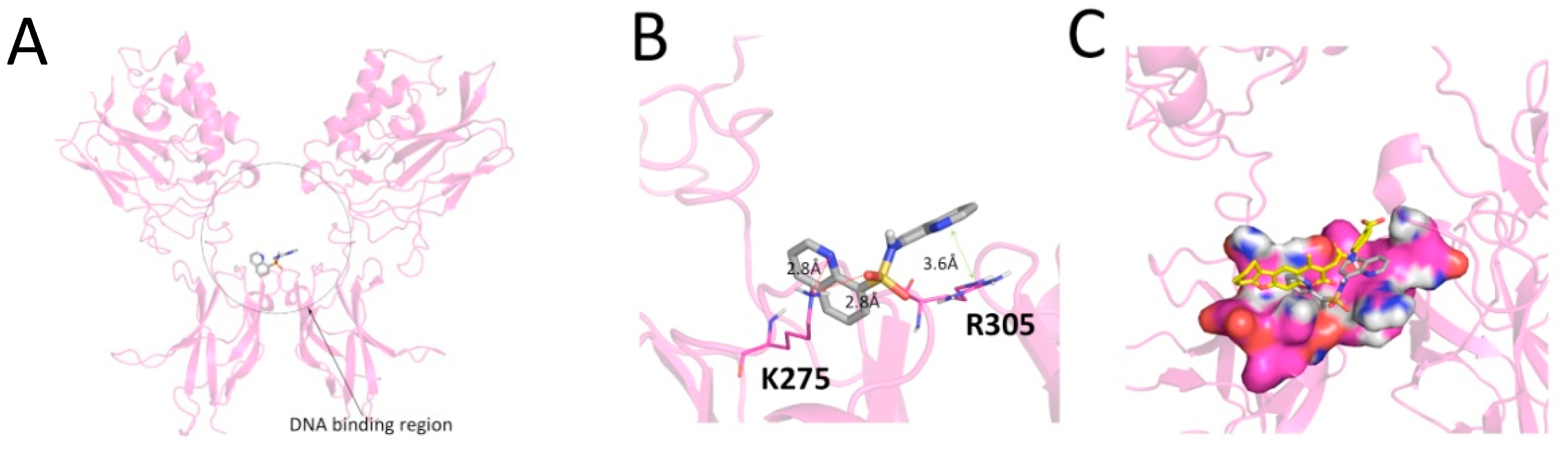
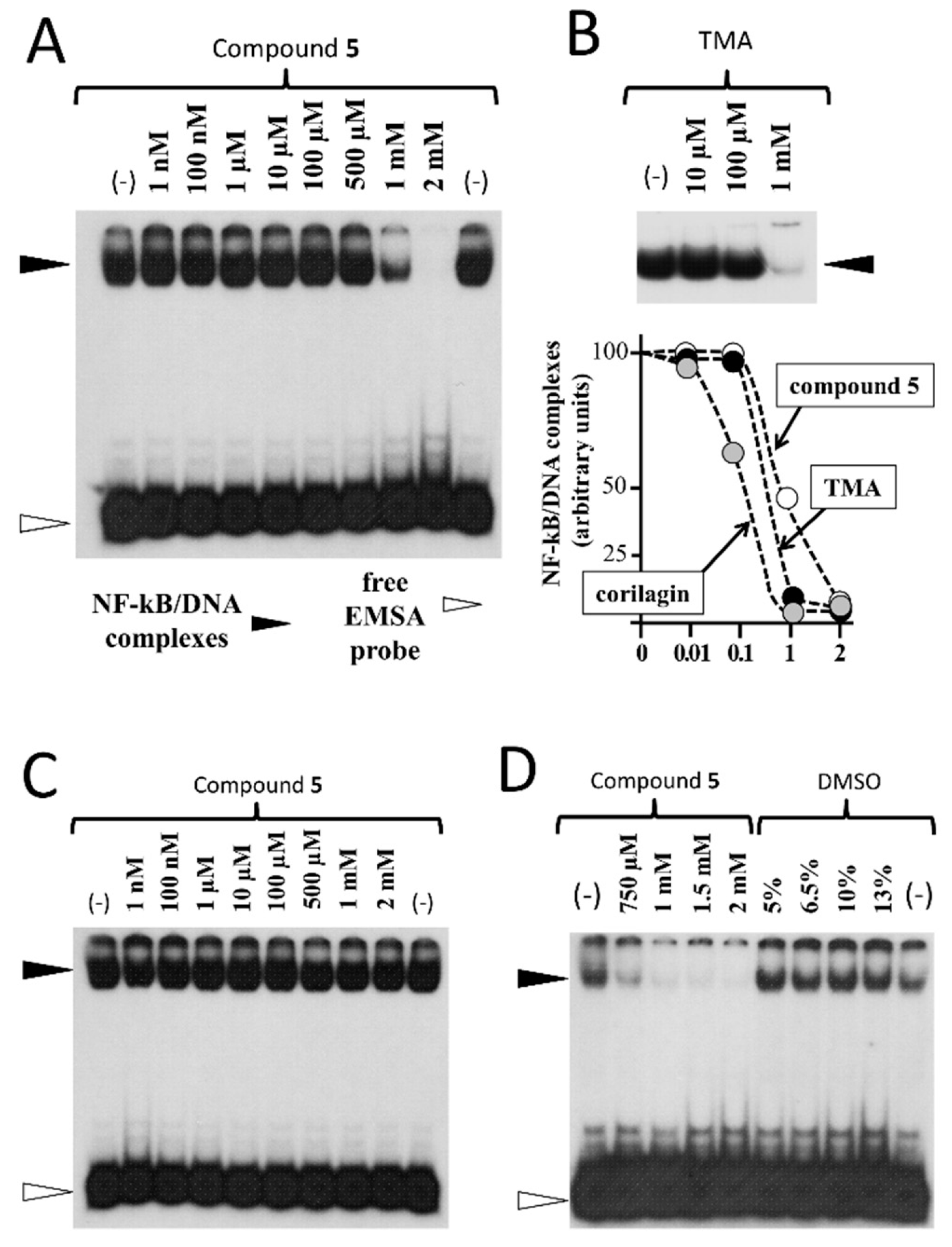
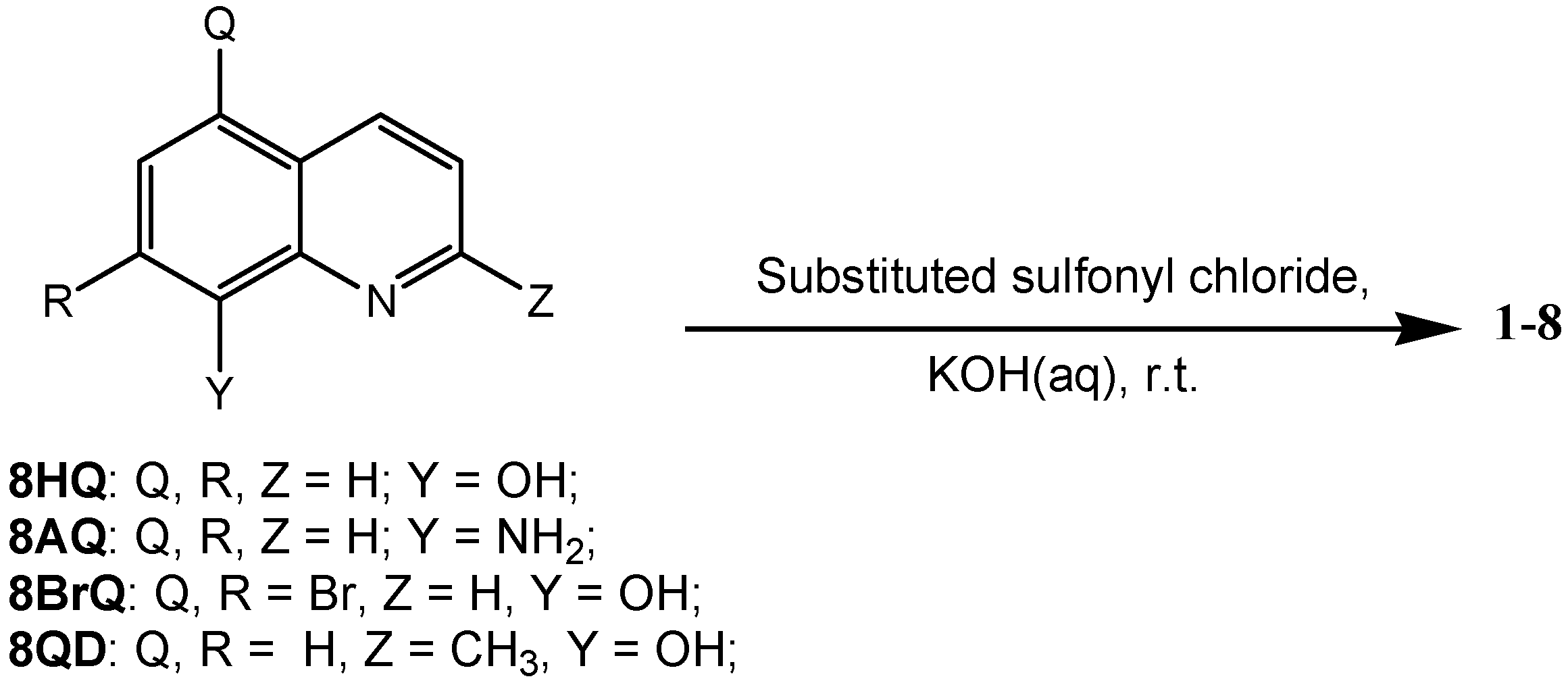
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials
3.1.2. General Procedure for the Synthesis of 1–4,6
3.1.3. General Procedure for the Synthesis of 5
3.1.4. General Procedure for the Synthesis of 8
3.1.5. General Procedure for the Synthesis of 7
3.2. Biological Analysis
3.2.1. Cell Lines and Cell Culture
3.2.2. In Vitro MTS Cytotoxicity Assay
3.2.3. Molecular Docking Analyses
3.2.4. Electrophoretic Mobility Shift Assay (EMSA)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Xu, J.Q.; Ward, E. Cancer Statistics, 2010. CA. Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Zhou, Y.; Chui, C.H.; Lam, K.H.; Law, S.; Chan, A.S.C.; Li, X.; Lam, A.K.Y.; Tang, J.C.O. Expression of insulin-like growth factor binding protein-5 (IGFBP5) reverses cisplatin-resistance in esophageal carcinoma. Cells 2018, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Tan, R.X.; Zhen, P.; Min, Z.D.; Iinuma, M.; Tanaka, T. Two steroidal alkaloid glycosides from Veratrum taliense. Phytochemistry 1990, 29, 359–361. [Google Scholar] [CrossRef]
- Ge, H.M.; Shen, Y.; Zhu, C.H.; Tan, S.H.; Ding, H.; Song, Y.C.; Tan, R.X. Penicidones A–C, three cytotoxic alkaloidal metabolites of an endophytic Penicillium sp. Phytochemistry 2008, 69, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Worthen, D.R.; Ghosheh, O.A.; Crooks, P.A. The in vitro anti-tumor activity of some crude and purified components of blackseed, Nigella sativa L. Anticancer Res. 1998, 18, 1527–1532. [Google Scholar] [PubMed]
- Fattorusso, E.; Taglialatela-Scafati, O. Modern Alkaloids: Structure, Isolation, Synthesis and Biology; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/9783527621071 (accessed on 20 September 2018).
- Kumar, S.; Bawa, S.; Gupta, H. Biological activities of quinoline derivatives. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, Z.; Hu, Y.; Wu, J.; Li, Y.; Huang, W. HZ08 reverse P-glycoprotein mediated multidrug resistance in vitro and in vivo. PLoS ONE 2015, 10, e0116886. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M. Alkaloids: Nature’s Curse or Blessing? Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Kouznetsov, V.V.M.; Leonor, Y.; Vargas, M.; Melendez Gomez, C.M. Recent progress in the synthesis of quinolines. Curr. Org. Chem. 2005, 9, 141–161. [Google Scholar] [CrossRef]
- Chung, P.-Y.; Bian, Z.-X.; Pun, H.-Y.; Chan, D.; Chan, A.S.-C.; Chui, C.-H.; Tang, J.C.-O.; Lam, K.-H. Recent advances in research of natural and synthetic bioactive quinolines. Future Med. Chem. 2015, 7, 947–967. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.H.; Lee, K.K.; Gambari, R.; Kok, S.H.; Kok, T.W.; Chan, A.S.; Bian, Z.X.; Wong, W.Y.; Wong, R.S.; Lau, F.Y.; et al. Anti-tumour and pharmacokinetics study of 2-Formyl-8-hydroxy-quinolinium chloride as galipea longiflora alkaloid analogue. Phytomedicine 2014, 21, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.-Y.; Gambari, R.; Chen, Y.-X.; Cheng, C.-H.; Bian, Z.-X.; Chan, A.S.-C.; Tang, J.C.-O.; Leung, P.H.-M.; Chui, C.-H.; Lam, K.-H. Development of 8-benzyloxy-substituted quinoline ethers and evaluation of their antimicrobial activities. Med. Chem. Res. 2014, 24, 1568–1577. [Google Scholar] [CrossRef]
- Ratheesh, M.; Sindhu, G.; Helen, A. Anti-inflammatory effect of quinoline alkaloid skimmianine isolated from Ruta Graveolens L. Inflamm. Res. 2013, 62, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Katanoda, K.; Matsuda, T. Five-year relative survival rate of liver cancer in the USA, Europe and Japan. Jpn. J. Clin. Oncol. 2014, 44, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Targeted therapy for liver cancer: Updated review in 2012. Curr. Cancer Drug Targets 2012, 12, 1062–1072. [Google Scholar] [PubMed]
- Lin, P.P.; Pang, Q.S.; Wang, P.; Lv, X.Y.; Liu, L.F.; Li, A.K. The targeted regulation of Gli1 by miR-361 to inhibit epithelia-mesenchymal transition and invasion of esophageal carcinoma cells. Cancer Biomark. 2018, 21, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput.-Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.; Vale, N.; de Freitas, V.; Moreira, R.; Mateus, N.; Gomes, P. Anti-tumoral activity of imidazoquines, a new class of antimalarials derived from primaquine. Bioorg. Med. Chem. Lett. 2009, 19, 6914–6917. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Álvarez, A.; Topala, T.; Estevan, F.; Sanz, F.; Alzuet-Piña, G. Photoinduced and self-activated nuclease activity of copper(II) complexes with N-(Quinolin-8-yl)quin-olin-8-sulfonamide—DNA and bovine serum albumin binding. Eur. J. Inorg. Chem. 2016, 2016, 982–994. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Shin, K.-J.; Lee, T.G.; Kim, E.; Lee, M.-S.; Ryu, S.H.; Suh, P.-G. G2 arrest and apoptosis by 2-amino-N-quinoline-8-yl-benzenesulfonamide (QBS), a novel cytotoxic compound. Biochem. Pharmacol. 2005, 69, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Borgatti, M.; Bezzerri, V.; Nicolis, E.; Lampronti, I.; Dechecchi, M.; Mancini, I.; Cabrini, G.; Saviano, M.; Avitabile, C.; et al. Effects of decoy molecules targeting NF-κB transcription factors in cystic fibrosis IB3–1 cells: Recruitment of NF-κB to the IL-8 gene promoter and transcription of the IL-8 gene. Artif. DNA PNA XNA 2012, 3, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Marzaro, G.; Guiotto, A.; Borgatti, M.; Finotti, A.; Gambari, R.; Breveglieri, G.; Chilin, A. Psoralen derivatives as inhibitors of NF-κB/DNA interaction: Synthesis, molecular modeling, 3D-QSAR, and biological evaluation. J. Med. Chem. 2013, 56, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Marzaro, G.; Lampronti, I.; D’Aversa, E.; Sacchetti, G.; Miolo, G.; Vaccarin, C.; Cabrini, G.; Dechecchi, M.C.; Gambari, R.; Chilin, A. Design, synthesis and biological evaluation of novel trimethylangelicin analogues targeting nuclear factor κB (NF-κB). Eur. J. Med. Chem. 2018, 151, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Lampronti, I.; Manzione, M.G.; Sacchetti, G.; Ferrari, D.; Spisani, S.; Bezzerri, V.; Finotti, A.; Borgatti, M.; Dechecchi, M.C.; Miolo, G.; et al. Differential effects of angelicin analogues on NF-κB activity and IL-8 gene expression in cystic fibrosis IB3-1 cells. Mediat. Inflamm. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Borgatti, M.; Lampronti, I.; Fabbri, E.; Brognara, E.; Bianchi, N.; Piccagli, L.; Yuen, C.W.M.; Kan, C.W.; Hau, D.K.P.; et al. Corilagin is a potent inhibitor of NF-κB activity and downregulates TNF-alpha induced expression of IL-8 gene in cystic fibrosis IB3-1 cells. Int. Immunopharmacol. 2012, 13, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Piccagli, L.; Fabbri, E.; Borgatti, M.; Bianchi, N.; Bezzerri, V.; Mancini, I.; Nicolis, E.; Dechecchi, C.M.; Lampronti, I.; Cabrini, G.; et al. Virtual screening against p50 NF-κB transcription factor for the identification of inhibitors of the NF-κB-DNA interaction and expression of NF-κB upregulated genes. ChemMedChem 2009, 4, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Neelgundmath, M.; Dinesh, K.R.; Mohan, C.D.; Li, F.; Dai, X.; Siveen, K.S.; Paricharak, S.; Mason, D.J.; Fuchs, J.E.; Sethi, G.; et al. Novel synthetic coumarins that targets NF-κB in hepatocellular carcinoma. Bioorg. Med. Chem. Lett. 2015, 25, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Shu, G.W.; Yue, L.; Zhao, W.H.; Xu, C.; Yang, J.; Wang, S.B.; Yang, X.Z. Isoliensinine, a bioactive alkaloid derived from embryos of nelumbo nucifera, induces hepatocellular carcinoma cell apoptosis through suppression of NF-κB signaling. J. Agric. Food. Chem. 2015, 63, 8793–8803. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Woo, K.J.; Lim, J.H.; Kim, S.; Lee, T.J.; Jung, E.M.; Lee, J.-M.; Park, J.-W.; Kwon, T.K. 8-Hydroxyquinoline inhibits iNOS expression and nitric oxide production by down-regulating LPS-induced activity of NF-κB and C/EBPβ in Raw 264.7 cells. Biochem. Biophys. Res. Commun. 2005, 329, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin. Drug Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Tang, J.; Cheng, C.H.; Bian, Z.X.; Wong, W.Y.; Lam, K.H.; Chui, C.H. Synthesis of hexahydrofuro [3,2-c]quinoline, a martinelline type analogue and investigation of its biological activity. SpringerPlus 2016, 5, 271. [Google Scholar] [CrossRef]
- Chopp, S.; Vanderwall, R.; Hult, A.; Klepser, M. Simeprevir and sofosbuvir for treatment of hepatitis C infection. Am. J. Health Syst. Pharm. 2015, 72, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M. Simeprevir: A review of its use in patients with chronic hepatitis C virus infection. Drugs 2015, 75, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, A.; Samuelsson, B.; Johansson, P.O.; Cummings, M.D.; Lenz, O.; Raboisson, P.; Simmen, K.; Vendeville, S.; de Kock, H.; Nilsson, M.; et al. Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J. Med. Chem. 2014, 57, 1673–1693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, Y.; Zhai, X.; Liu, X.; Sun, L.; Ren, Y.; Gong, P. Synthesis and anti-HBV activities evaluation of new ethyl 8-Imidazolylmethyl-7-hydroxyquinoline-3-carboxylate derivatives in vitro. Arch. Pharm. 2008, 341, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Zhai, X.; Li, T.; Liu, D.; Zhang, T.; Wang, B.; Gong, P. Design, synthesis and antiproliferative activity of novel 2-substituted-4-amino-6-halogenquinolines. Molecules 2012, 17, 5870–5881. [Google Scholar] [CrossRef] [PubMed]
- Rouffet, M.; de Oliveira, C.A.F.; Udi, Y.; Agrawal, A.; Sagi, I.; McCammon, J.A.; Cohen, S.M. From sensors to silencers: Quinoline-and benzimidazole-sulfonamides as inhibitors for zinc proteases. J. Am. Chem. Soc. 2010, 132, 8232–8233. [Google Scholar] [CrossRef] [PubMed]
- Arepalli, S.K.; Choi, M.; Jung, J.K.; Lee, H. Novel NF-κB inhibitors: A patent review (2011–2014). Expert Opin. Ther. Patents 2015, 25, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Castranova, V.; Shi, X. New insights into the role of fuclear factor-κB in cell growth regulation. Am. J. Pathol. 2001, 159, 387–397. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Gurumurthy, S.; Rangnekar, V.M. Suppression of PTEN expression by NF-κB prevents apoptosis. Mol. Cell. Biol. 2004, 24, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Tian, L.L.; Khan, M.N.; Zhang, L.; Chen, Q.; Zhao, Y.; Yan, Q.; Fu, L.; Liu, J.W. Ginsenoside Rg3 sensitizes hypoxic lung cancer cells to cisplatin via blocking of NF-κB mediated epithelial-mesenchymal transition and sternness. Cancer Lett. 2018, 415, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Pyo, M.J.; Bae, S.K.; Heo, Y.; Choudhary, I.; Hwang, D.; Yang, H.; Kim, J.H.; Chae, J.; Han, C.H.; et al. Nemopilema nomurai jellyfish venom exerts an anti-metastatic effect by inhibiting Smad- and NF-κB-mediated epithelial-mesenchymal transition in HepG2 cells. Sci. Rep. 2018, 8, 2808. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF–ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Mu, G.G.; Ding, Q.S.; Li, Y.X.; Shi, Y.B.; Dai, J.F.; Yu, H.G. Phosphatase and tensin homolog (PTEN) represses colon cancer progression through inhibiting paxillin transcription via PI3K/AKT/NF-κB Pathway. J. Biol. Chem. 2015, 290, 15018–15029. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Song, L.; Gong, H.; Liu, A.; Lin, X.; Wu, J.; Li, M.; Li, J. Nkx2-8 downregulation promotes angiogenesis and activates NF-κB in esophageal cancer. Cancer Res. 2013, 73, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.-P.; Liu, S.-K.; Li, Z.-J.; Li, B.; Zhou, Y.-L.; Guo, R.; Huang, L.-H. NF-κB p65 modulates the telomerase reverse transcriptase in the HepG2 hepatoma cell line. Eur. J. Pharmacol. 2011, 672, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Chui, C.H.; Chan, S.W.; Kok, S.H.L.; Chan, D.; Tsoi, M.Y.T.; Leung, P.H.M.; Lam, A.K.Y.; Chan, A.S.C.; Lam, K.H.; et al. Synthesis of 8-Hydroxyquinoline derivatives as novel antitumor agents. ACS Med. Chem. Lett. 2013, 4, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.C.O.; Wan, T.S.K.; Wong, N.; Pang, E.; Lam, K.Y.; Law, S.Y.; Chow, L.M.C.; Ma, E.S.K.; Chan, L.C.; Wong, J.; et al. Establishment and characterization of a new xenograft-derived human esophageal squamous cell carcinoma cell line SLMT-1 of Chinese origin. Cancer Genet. Cytogenet. 2001, 124, 36–41. [Google Scholar] [CrossRef]
- Pun, I.H.Y.; Chan, D.; Chan, S.H.; Chung, P.Y.; Zhou, Y.Y.; Law, S.; Lam, A.K.Y.; Chui, C.H.; Chan, A.S.C.; Lam, K.H.; et al. Anti-cancer effects of a novel quinoline derivative 83b1 on human esophageal squamous cell carcinoma through down-regulation of COX-2 mRNA and PGE(2). Cancer Res. Treat. 2017, 49, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

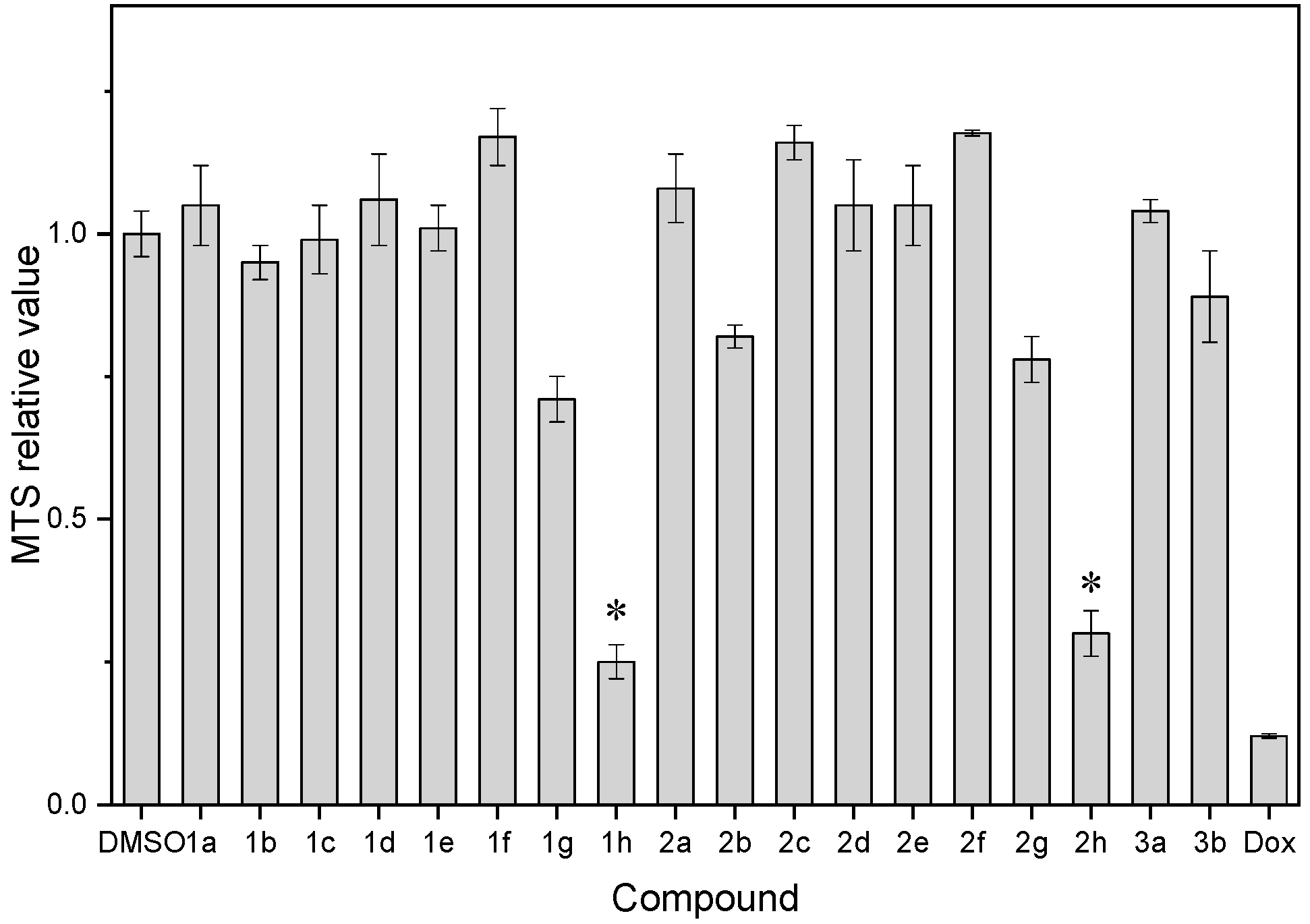
| Compound | MTS Relative Values a (Mean ± SD) | Log P b | Drug Score c | Nuclear Factor NF-κB p65 Subunit: E-Value d |
|---|---|---|---|---|
| 1a | 1.05 ± 0.07 | 2.76 | 0.15 | 2.59 × 10−20 |
| 1b | 0.95 ± 0.03 | 4.10 | 0.37 | 4.01 × 10−21 |
| 1c | 0.99 ± 0.06 | 3.17 | 0.36 | 1.12 × 10−16 |
| 1d | 1.06 ± 0.08 | 3.19 | 0.38 | 2.38 × 10−12 |
| 1e | 1.01 ± 0.04 | 3.50 | 0.22 | 7.92 × 10−13 |
| 1f | 1.17 ± 0.05 | 3.50 | 0.36 | 7.92 × 10−13 |
| 1g | 0.71 ± 0.04 | 3.21 | 0.21 | 5.39 × 10−17 |
| 1h | 0.25 ± 0.03 | 3.27 | 0.10 | 8.61 × 10−7 |
| 2a | 1.08 ± 0.06 | 3.18 | 0.15 | 6.20 × 10−17 |
| 2b | 0.82 ± 0.02 | 3.59 | 0.33 | 9.52 × 10−18 |
| 2c | 1.16 ± 0.03 | 3.60 | 0.33 | 1.13 × 10−20 |
| 2d | 1.05 ± 0.08 | 3.61 | 0.34 | 1.01 × 10−15 |
| 2e | 1.05 ± 0.07 | 3.91 | 0.20 | 2.74 × 10−21 |
| 2f | 1.18 ± 0.005 | 3.90 | 0.33 | 1.46 × 10−18 |
| 2g | 0.78 ± 0.04 | 3.63 | 0.19 | 4.07 × 10−16 |
| 2h | 0.30 ± 0.04 | 3.66 | 0.09 | 7.92 × 10−13 |
| 3a | 1.04 ± 0.02 | 3.12 | 0.22 | 1.08 × 10−17 |
| 3b | 0.89 ± 0.08 | 3.49 | 0.20 | 7.19 × 10−15 |

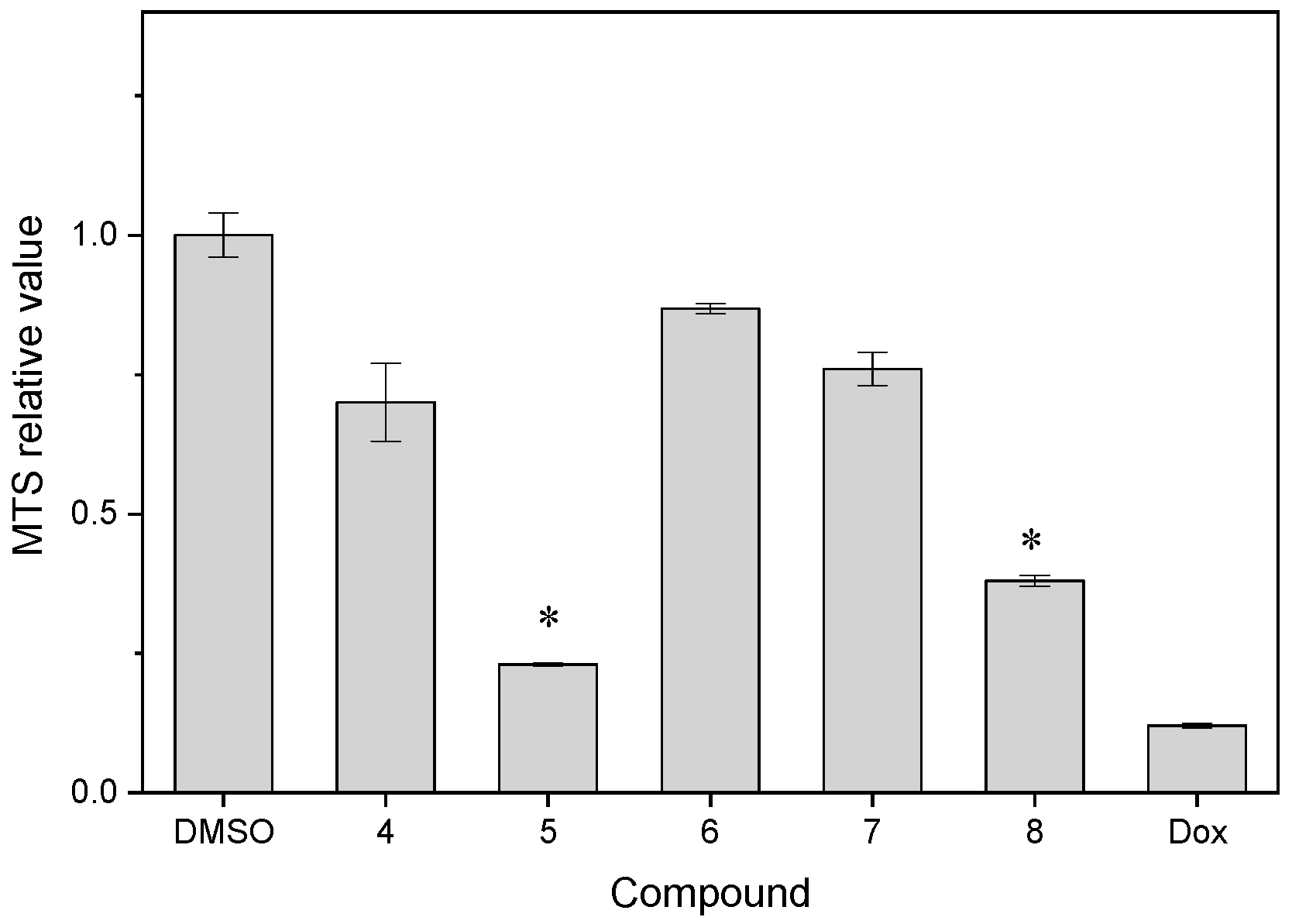
| Compound | MTS Relative Values a (Mean ± SD) | Log P b | Drug Score c | Nuclear Factor NF-κB p65 Subunit: E-Value d |
|---|---|---|---|---|
| 4 | 0.70 ± 0.07 | 2.60 | 0.45 | 4.63 × 10−24 |
| 5 | 0.230 ± 0.003 | 2.40 | 0.60 | 9.05 × 10−71 |
| 6 | 0.868 ± 0.009 | 3.04 | 0.41 | 4.45 × 10−20 |
| 7 | 0.76 ± 0.03 | 2.96 | 0.37 | 1.27 × 10−36 |
| 8 | 0.38 ± 0.01 | 3.66 | 0.13 | 5.18 × 10−9 |
| Cell Line | MTS50 Values (μM) (Mean ± SD) | |
|---|---|---|
| 5 | Dox | |
| Hep3B | 2.91 ± 0.09 | 0.27 ± 0.02 |
| SLMT-1 | 6.53 ± 0.06 | 0.41 ± 0.03 |
| MCF-7 | 1.23 ± 0.03 | 0.28 ± 0.04 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, P.Y.; Lam, P.L.; Zhou, Y.; Gasparello, J.; Finotti, A.; Chilin, A.; Marzaro, G.; Gambari, R.; Bian, Z.; Kwok, W.M.; et al. Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma. Cells 2018, 7, 177. https://doi.org/10.3390/cells7100177
Chung PY, Lam PL, Zhou Y, Gasparello J, Finotti A, Chilin A, Marzaro G, Gambari R, Bian Z, Kwok WM, et al. Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma. Cells. 2018; 7(10):177. https://doi.org/10.3390/cells7100177
Chicago/Turabian StyleChung, Po Yee, Pik Ling Lam, Yuanyuan Zhou, Jessica Gasparello, Alessia Finotti, Adriana Chilin, Giovanni Marzaro, Roberto Gambari, Zhaoxiang Bian, Wai Ming Kwok, and et al. 2018. "Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma" Cells 7, no. 10: 177. https://doi.org/10.3390/cells7100177
APA StyleChung, P. Y., Lam, P. L., Zhou, Y., Gasparello, J., Finotti, A., Chilin, A., Marzaro, G., Gambari, R., Bian, Z., Kwok, W. M., Wong, W. Y., Wang, X., Lam, A. K.-y., Chan, A. S.-c., Li, X., Ma, J. Y. W., Chui, C. H., Lam, K. H., & Tang, J. C. O. (2018). Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma. Cells, 7(10), 177. https://doi.org/10.3390/cells7100177