Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis

Abstract
{kind=link}
{kind=link}
{kind=link}
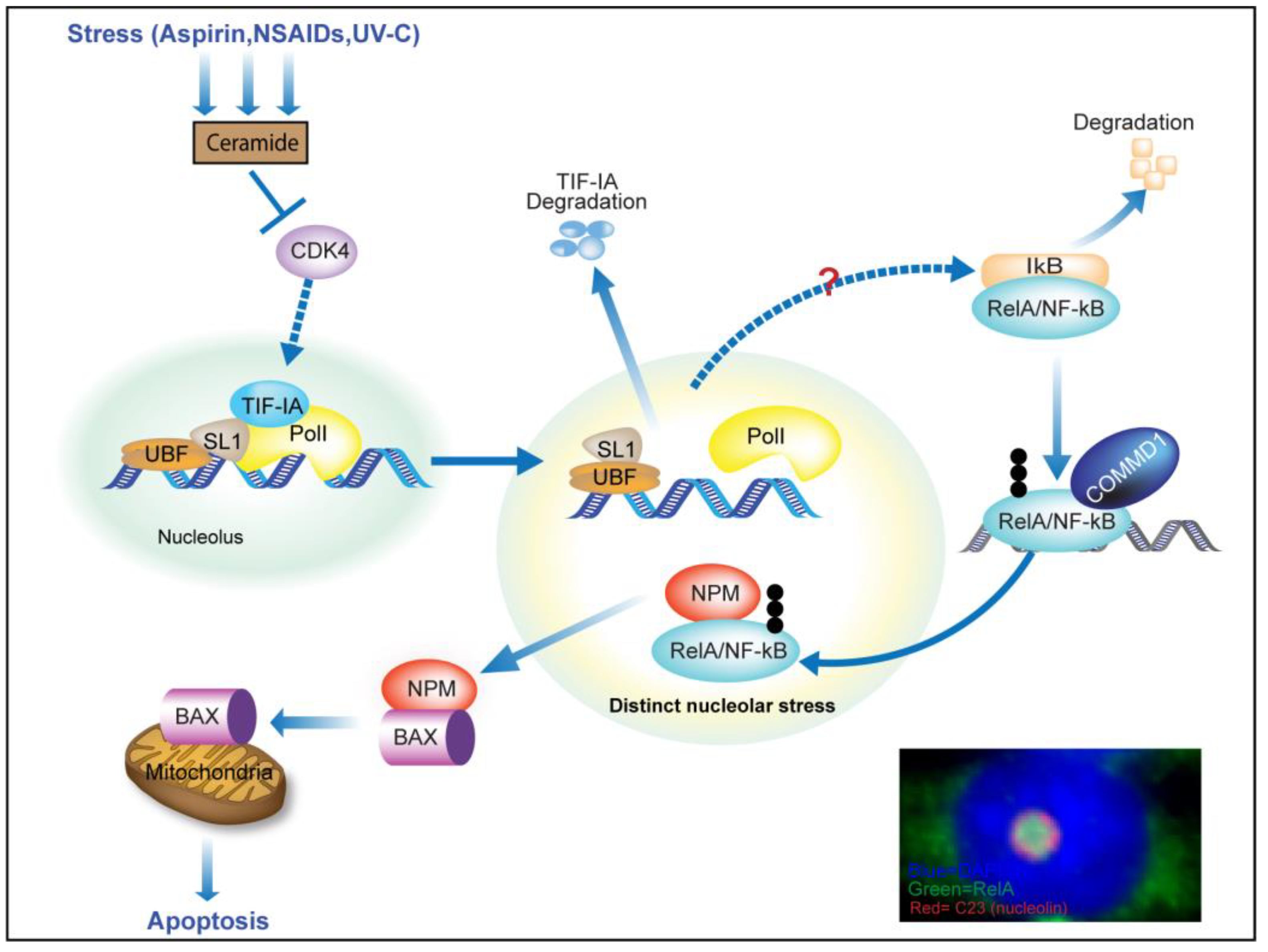{kind=link}
1. Introduction
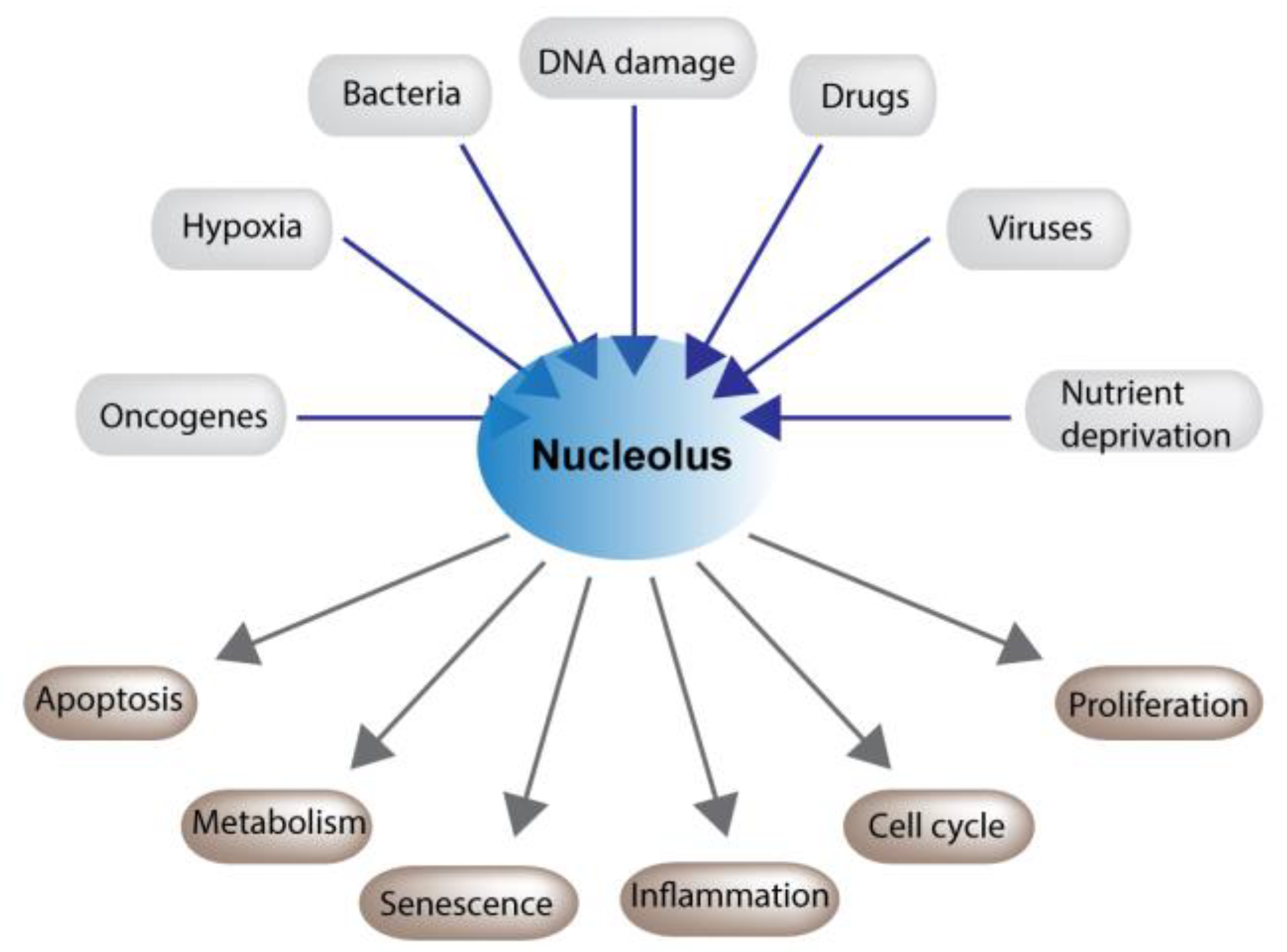
2. The Nucleolus and Stress
2.1. Nucleolar Maintenance of Cell Physiology
2.2. p53 Dependent and Independent Consequences of Nucleolar Stress
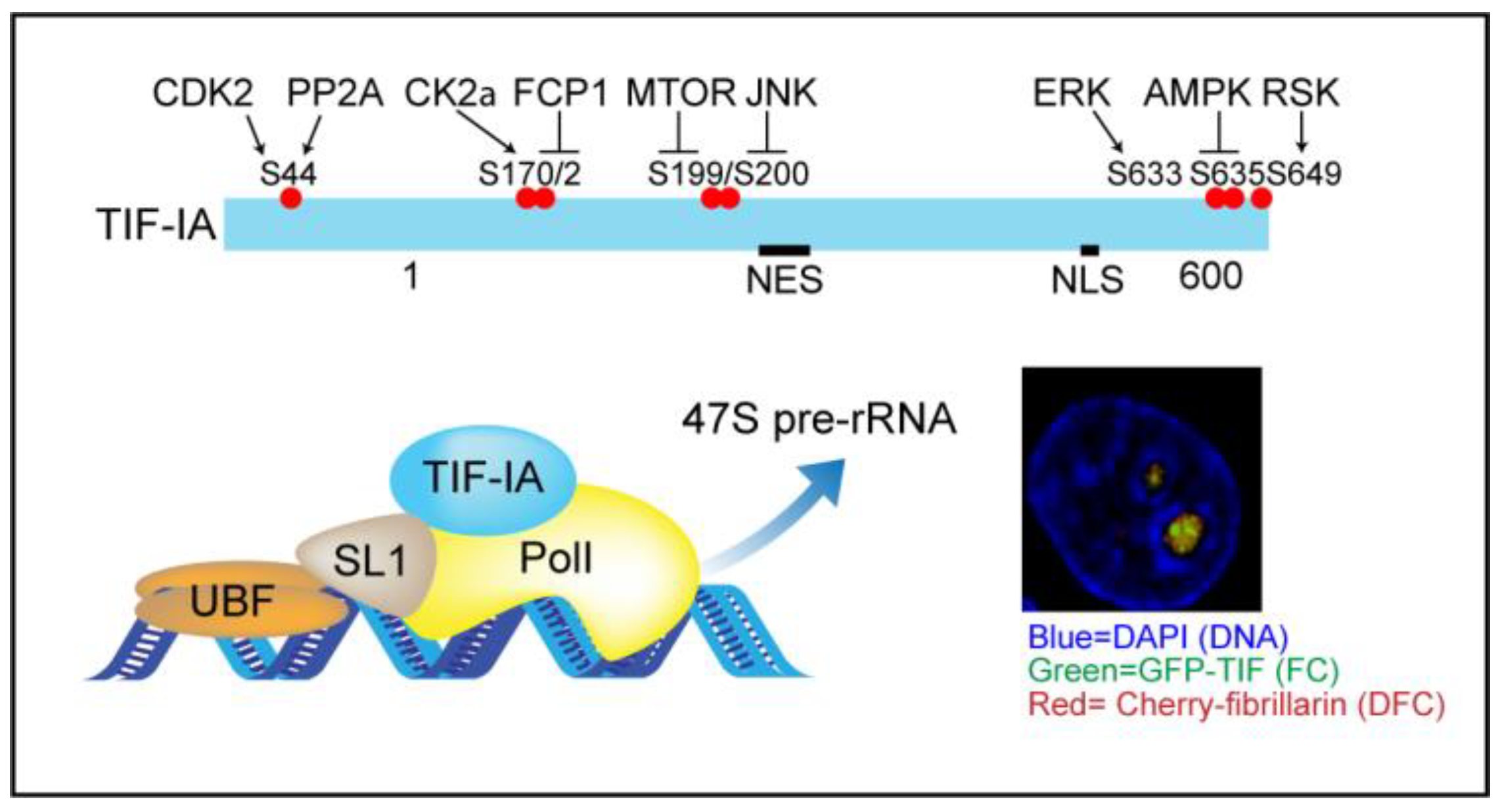
3. TIF-IA-NF-κB Nucleolar Stress
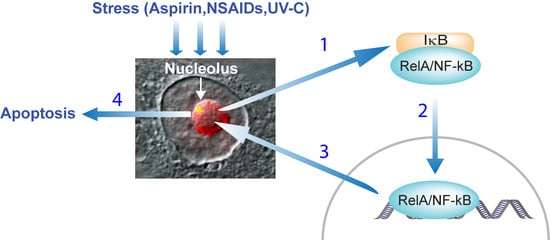
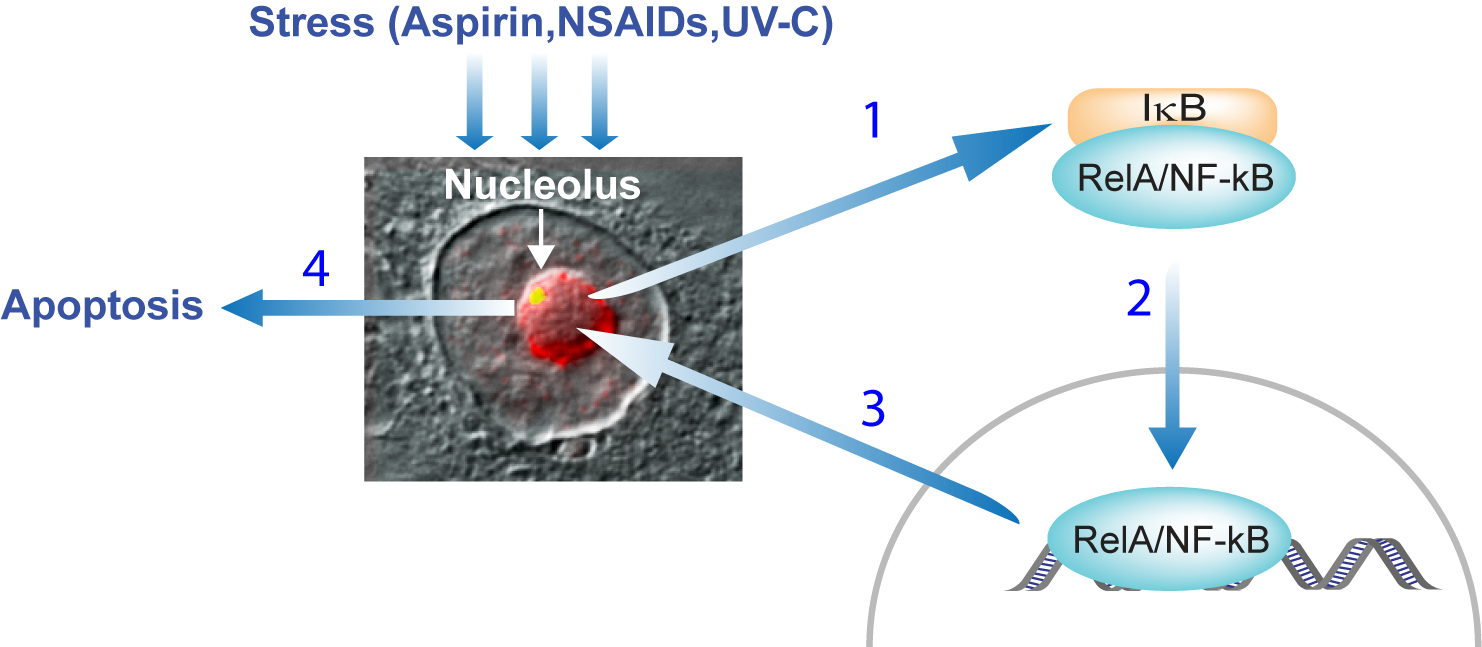
3.1. Stress Activation of the NF-κB Pathway
3.2. TIF-IA Degradation—A Novel Form of Nucleolar Stress
3.3. Nucleolar Enlargement as a Consequence of TIF-IA Degradation
3.4. Activation of the NF-κB Pathway as a Consequence of TIF-IA Degradation
3.5. TIF-IA-NF-κB Nucleolar Stress and the Induction of Apoptosis
4. Nucleolar Sequestration of RelA and Apoptosis
5. Therapeutic Relevance of Crosstalk between Nucleoli and the NF-κB Pathway
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Activators and target genes of rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lobb, I.T.; Morin, P.; Novo, S.M.; Simpson, J.; Kennerknecht, K.; von Kriegsheim, A.; Batchelor, E.E.; Oakley, F.; Stark, L.A. Identification of a novel TIF-IA-nf-κB nucleolar stress response pathway. Nucleic Acids Res. 2018, 46, 6188–6205. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Dunlop, M.G. Nucleolar sequestration of rela (p65) regulates nf-κB-driven transcription and apoptosis. Mol. Cell. Biol. 2005, 25, 5985–6004. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, N.; Simpson, J.; Taylor, G.; Rafique, S.; Whitehouse, A.; Hiscox, J.; Stark, L.A. Nucleolar nf-κB/rela mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 2011, 18, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Schnapp, A.; Pfleiderer, C.; Rosenbauer, H.; Grummt, I. A growth-dependent transcription initiation factor (TIF-IA) interacting with rna polymerase i regulates mouse ribosomal rna synthesis. EMBO J. 1990, 9, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhao, J.; Zentgraf, H.; Hoffmann-Rohrer, U.; Grummt, I. Multiple interactions between RNA polymerase I, TIF-IA and TAF(I) subunits regulate preinitiation complex assembly at the ribosomal gene promoter. EMBO Rep. 2002, 3, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zhou, W. TIF-IA: An oncogenic target of pre-ribosomal RNA synthesis. Biochim. Biophys. Acta 2016, 1866, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.E.; Mais, C.; Prieto, J.L.; McStay, B. A role for upstream binding factor in organizing ribosomal gene chromatin. Biochem. Soc. Symp. 2006, 77–84. [Google Scholar] [CrossRef]
- Mangan, H.; Gailin, M.O.; McStay, B. Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 2017, 284, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.T.; Elbaum-Garfinkle, S.; Holehouse, A.S.; Chen, C.C.; Feric, M.; Arnold, C.B.; Priestley, R.D.; Pappu, R.V.; Brangwynne, C.P. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 2017, 9, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.Y.; Pederson, T. Connecting the nucleolus to the cell cycle and human disease. FASEB J. 2014, 28, 3290–3296. [Google Scholar] [CrossRef] [PubMed]
- Nunez Villacis, L.; Wong, M.S.; Ferguson, L.L.; Hein, N.; George, A.J.; Hannan, K.M. New roles for the nucleolus in health and disease. Bioessays 2018, 40, e1700233. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman, H.E.D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2016, 8, 16083. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Grummt, I. Dynamic regulation of nucleolar architecture. Curr. Opin. Cell Biol. 2018, 52, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.D.; Audas, T.E.; Uniacke, J.; Trinkle-Mulcahy, L.; Lee, S. Environmental cues induce a long noncoding RNA-dependent remodeling of the nucleolus. Mol. Biol. Cell 2013, 24, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Cellular stress and nucleolar function. Cell Cycle 2005, 4, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Holmberg Olausson, K.; Nister, M.; Lindstrom, M.S. P53 -dependent and -independent nucleolar stress responses. Cells 2012, 1, 774–798. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.M.; Bai, B.; Boisvert, F.M.; Latonen, L.; Rantanen, V.; Simpson, J.C.; Pepperkok, R.; Lamond, A.I.; Laiho, M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to uv and ionizing radiation. Mol. Cell. Proteom. 2011, 10, M111. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Lam, Y.W.; Lamont, D.; Lamond, A.I. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteom. 2010, 9, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.J.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim. Biophys. Acta 2015, 1849, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Russo, G. Ribosomal proteins control or bypass p53 during nucleolar stress. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Al Baker, E.A.; Boyle, J.; Harry, R.; Kill, I.R. A p53-independent pathway regulates nucleolar segregation and antigen translocation in response to DNA damage induced by uv irradiation. Exp. Cell Res. 2004, 292, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Brighenti, E.; Vici, M.; Mazzini, G.; Trere, D.; Montanaro, L.; Derenzini, M. Selective inhibition of rRNA transcription downregulates E2F-1: A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 2011, 124, 3017–3028. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Esposito, D.; Catillo, M.; Pietropaolo, C.; Crescenzi, E.; Russo, G. Human rpL3 induces G1/S arrest or apoptosis by modulating p21waf1/cip1 levels in a p53-independent manner. Cell Cycle 2013, 12, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Iadevaia, V.; Caldarola, S.; Biondini, L.; Gismondi, A.; Karlsson, S.; Dianzani, I.; Loreni, F. Pim1 kinase is destabilized by ribosomal stress causing inhibition of cell cycle progression. Oncogene 2010, 29, 5490–5499. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A. Pro-inflammatory signaling: Last pieces in the NF-κB puzzle? Curr. Biol. 1998, 8, R19–R22. [Google Scholar] [CrossRef]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The k48-k63 branched ubiquitin chain regulates NF-κB signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Miyamoto, S. Many faces of NF-κB signaling induced by genotoxic stress. J. Mol. Med. 2007, 85, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 is a C-terminal IκB kinase responsible for NF-κB activation during the uv response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, R.C. GCN2 phosphorylation of eIF2α activates NF-κB in response to UV irradiation. Biochem. J. 2005, 385, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. Mtor-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Parlato, R.; Bierhoff, H. Role of nucleolar dysfunction in neurodegenerative disorders: A game of genes. AIMS Mol. Sci. 2015, 2, 211–224. [Google Scholar] [CrossRef]
- Szymanski, J.; Mayer, C.; Hoffmann-Rohrer, U.; Kalla, C.; Grummt, I.; Weiss, M. Dynamic subcellular partitioning of the nucleolar transcription factor TIF-IA under ribotoxic stress. Biochim. Biophys. Acta 2009, 1793, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Bierhoff, H.; Grummt, I. The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rrna synthesis. Genes Dev. 2005, 19, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Grone, H.J.; Schutz, G.; Grummt, I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Parlato, R.; Kreiner, G.; Erdmann, G.; Rieker, C.; Stotz, S.; Savenkova, E.; Berger, S.; Grummt, I.; Schutz, G. Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J. Neurosci. 2008, 28, 12759–12764. [Google Scholar] [CrossRef] [PubMed]
- Fatyol, K.; Grummt, I. Proteasomal atpases are associated with rdna: The ubiquitin proteasome system plays a direct role in RNA polymerase i transcription. Biochim. Biophys. Acta 2008, 1779, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Bailly, A.; Perrin, A.; Bou Malhab, L.J.; Pion, E.; Larance, M.; Nagala, M.; Smith, P.; O’Donohue, M.F.; Gleizes, P.E.; Zomerdijk, J.; et al. The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene 2016, 35, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Bierhoff, H.; Dundr, M.; Michels, A.A.; Grummt, I. Phosphorylation by casein kinase 2 facilitates rrna gene transcription by promoting dissociation of TIF-IA from elongating rna polymerase i. Mol. Cell. Biol. 2008, 28, 4988–4998. [Google Scholar] [CrossRef] [PubMed]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2α coordinates rrna transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed]
- Birbach, A.; Bailey, S.T.; Ghosh, S.; Schmid, J.A. Cytosolic, nuclear and nucleolar localization signals determine subcellular distribution and activity of the NF-κB inducing kinase nik. J. Cell Sci. 2004, 117, 3615–3624. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein s3: A kh domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Maiolino, S.; Pagliara, V.; Ungaro, F.; Tatangelo, F.; Leone, A.; Scalia, G.; Budillon, A.; Quaglia, F.; Russo, G. Enhancement of 5-FU sensitivity by the proapoptotic rpl3 gene in p53 null colon cancer cells through combined polymer nanoparticles. Oncotarget 2016, 7, 79670–79687. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Kamalakaran, S. Pro-apoptotic role of NF-κB: Implications for cancer therapy. Biochim. Biophys. Acta 2006, 1766, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Din, F.V.N.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NF-κB signalling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Rocha, S.; Perkins, N.D. Active repression of antiapoptotic gene expression by rela(p65) NF-κB. Mol. Cell 2004, 13, 853–865. [Google Scholar] [CrossRef]
- Fillet, M.; Bentires-Alj, M.; Deregowski, V.; Greimers, R.; Gielen, J.; Piette, J.; Bours, V.; Merville, M.P. Mechanisms involved in exogenous C2- and C6-ceramide-induced cancer cell toxicity. Biochem. Pharmacol. 2003, 65, 1633–1642. [Google Scholar] [CrossRef]
- Loveridge, C.J.; Macdonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the rela(p65) subunit of NF-κB. Oncogene 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Stark, L.A.; Dunlop, M.G.; Clarke, A.R. Suppression of intestinal and mammary neoplasia by lifetime administration of aspirin in ApcMin/+ and Apcmin/+, Msh2-/- mice. Cancer Res. 2001, 61, 7060–7064. [Google Scholar] [PubMed]
- Salmina, K.; Huna, A.; Inashkina, I.; Belyayev, A.; Krigerts, J.; Pastova, L.; Vazquez-Martin, A.; Erenpreisa, J. Nucleolar aggresomes mediate release of pericentric heterochromatin and nuclear destruction of genotoxically treated cancer cells. Nucleus 2017, 8, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Nucleolar aggresomes as counterparts of cytoplasmic aggresomes in proteotoxic stress. Proteasome inhibitors induce nuclear ribonucleoprotein inclusions that accumulate several key factors of neurodegenerative diseases and cancer. Bioessays 2011, 33, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Jacob, M.D.; Lee, S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 2012, 45, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Rossi, A.; Riccio, A.; Trotta, E.; Santoro, M.G. Human NF-κB repressing factor acts as a stress-regulated switch for ribosomal rna processing and nucleolar homeostasis surveillance. Proc. Natl. Acad. Sci. USA 2017, 114, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. Non-activated p53 co-localizes with sites of transcription within both the nucleoplasm and the nucleolus. Oncogene 2000, 19, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Moore, H.M.; Bai, B.; Jaamaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Jacob, M.D.; Lee, S. The nucleolar detention pathway: A cellular strategy for regulating molecular networks. Cell Cycle 2012, 11, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- Cancer Registration Statistics Scotland 1986–1995; ISD: Scotland, UK, 1998.
- Parrondo, R.; de las Pozas, A.; Reiner, T.; Rai, P.; Perez-Stable, C. NF-κB activation enhances cell death by antimitotic drugs in human prostate cancer cells. Mol. Cancer 2010, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Forscher, C.; Di Vizio, D.; Koeffler, H.P. Induction of p53-independent apoptosis by ectopic expression of HOXA5 in human liposarcomas. Sci. Rep. 2015, 5, 12580. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. P38-mediated inactivation of cyclin d1/cyclin-dependent kinase 4 stimulates nucleolar translocation of rela and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. Cdk4 inhibitors and apoptosis: A novel mechanism requiring nucleolar targeting of rela. Cell Cycle 2007, 6, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Loveridge, C.J.; Simpson, J.; Clipson, A.; Reinhardt, K.; Dunlop, M.G.; Stark, L.A. Nucleolar targeting of rela(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010, 70, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Dadsetan, S.; Balzano, T.; Forteza, J.; Agusti, A.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Hernandez-Rabaza, V.; Gomez-Gimenez, B.; ElMlili, N.; Llansola, M.; et al. Infliximab reduces peripheral inflammation, neuroinflammation, and extracellular gaba in the cerebellum and improves learning and motor coordination in rats with hepatic encephalopathy. J. Neuroinflamm. 2016, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.; Simpson, J.; Morin, P.; Loveridge, C.J.; Williams, A.C.; Novo, S.M.; Stark, L.A. P300-mediated acetylation of COMMD1 regulates its stability, and the ubiquitylation and nucleolar translocation of the rela NF-κB subunit. J. Cell Sci. 2014, 127, 3659–3665. [Google Scholar] [CrossRef] [PubMed]
- Kerr, L.E.; Birse-Archbold, J.L.; Short, D.M.; McGregor, A.L.; Heron, I.; Macdonald, D.C.; Thompson, J.; Carlson, G.J.; Kelly, J.S.; McCulloch, J.; et al. Nucleophosmin is a novel bax chaperone that regulates apoptotic cell death. Oncogene 2007, 26, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Finlayson, K.; Salvo-Chirnside, E.; MacDonald, D.; McCulloch, J.; Kerr, L.; Sharkey, J. Characterisation of the bax-nucleophosmin interaction: The importance of the bax C-terminus. Apoptosis 2008, 13, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Kreiner, G.; Bierhoff, H.; Armentano, M.; Rodriguez-Parkitna, J.; Sowodniok, K.; Naranjo, J.R.; Bonfanti, L.; Liss, B.; Schutz, G.; Grummt, I.; et al. A neuroprotective phase precedes striatal degeneration upon nucleolar stress. Cell Death Differ. 2013, 20, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Evsyukov, V.; Domanskyi, A.; Bierhoff, H.; Gispert, S.; Mustafa, R.; Schlaudraff, F.; Liss, B.; Parlato, R. Genetic mutations linked to parkinson′s disease differentially control nucleolar activity in pre-symptomatic mouse models. Dis. Models Mech. 2017, 10, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernandez, O.; Freije, J.M.; Lopez-Otin, C. NF-κB signaling as a driver of ageing. Int. Rev. Cell Mol. Biol. 2016, 326, 133–174. [Google Scholar] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Barnetson, R.A.; Cetnarskyj, R.; Stark, L.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Effect of aspirin and nsaids on risk and survival from colorectal cancer. Gut 2010, 59, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Thorat, M.A.; Bosetti, C.; Brown, P.H.; Burn, J.; Cook, N.R.; Ford, L.G.; Jacobs, E.J.; Jankowski, J.A.; La, V.C.; et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the capp2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-inflammatory drugs and risk of alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimers Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Roy, A.; Kundu, M.; Jana, M.; Luan, C.H.; Gonzalez, F.J.; Pahan, K. Aspirin binds to pparalpha to stimulate hippocampal plasticity and protect memory. Proc. Natl. Acad. Sci. USA 2018, 115, E7408–E7417. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase i with an oral small molecule cx-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Frank, D.; Son, J.; Hannan, K.M.; Hannan, R.D.; Chan, K.T.; Pearson, R.B.; Sanij, E. The potential of targeting ribosome biogenesis in high-grade serous ovarian cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of rna polymerase i that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting cdk4 and cdk6: From discovery to therapy. Cancer Discov. 2015, 6, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Myant, K.; Reed, K.R.; Ridgway, R.A.; Athineos, D.; van den Brink, G.R.; Muncan, V.; Clevers, H.; Clarke, A.R.; Sicinski, P.; et al. Cyclin d2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following apc loss. Cancer Res. 2010, 70, 8149–8158. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Dunlop, M.G.; Stark, L.A. Evidence for colorectal cancer cell specificity of aspirin effects on nf kappa b signalling and apoptosis. Br. J. Cancer 2004, 91, 381–388. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Stark, L.A. Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis. Cells 2018, 7, 157. https://doi.org/10.3390/cells7100157
Chen J, Stark LA. Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis. Cells. 2018; 7(10):157. https://doi.org/10.3390/cells7100157
Chicago/Turabian StyleChen, Jingyu, and Lesley A. Stark. 2018. "Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis" Cells 7, no. 10: 157. https://doi.org/10.3390/cells7100157
APA StyleChen, J., & Stark, L. A. (2018). Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis. Cells, 7(10), 157. https://doi.org/10.3390/cells7100157