Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanism for Activation of EGFR by Ligand Binding
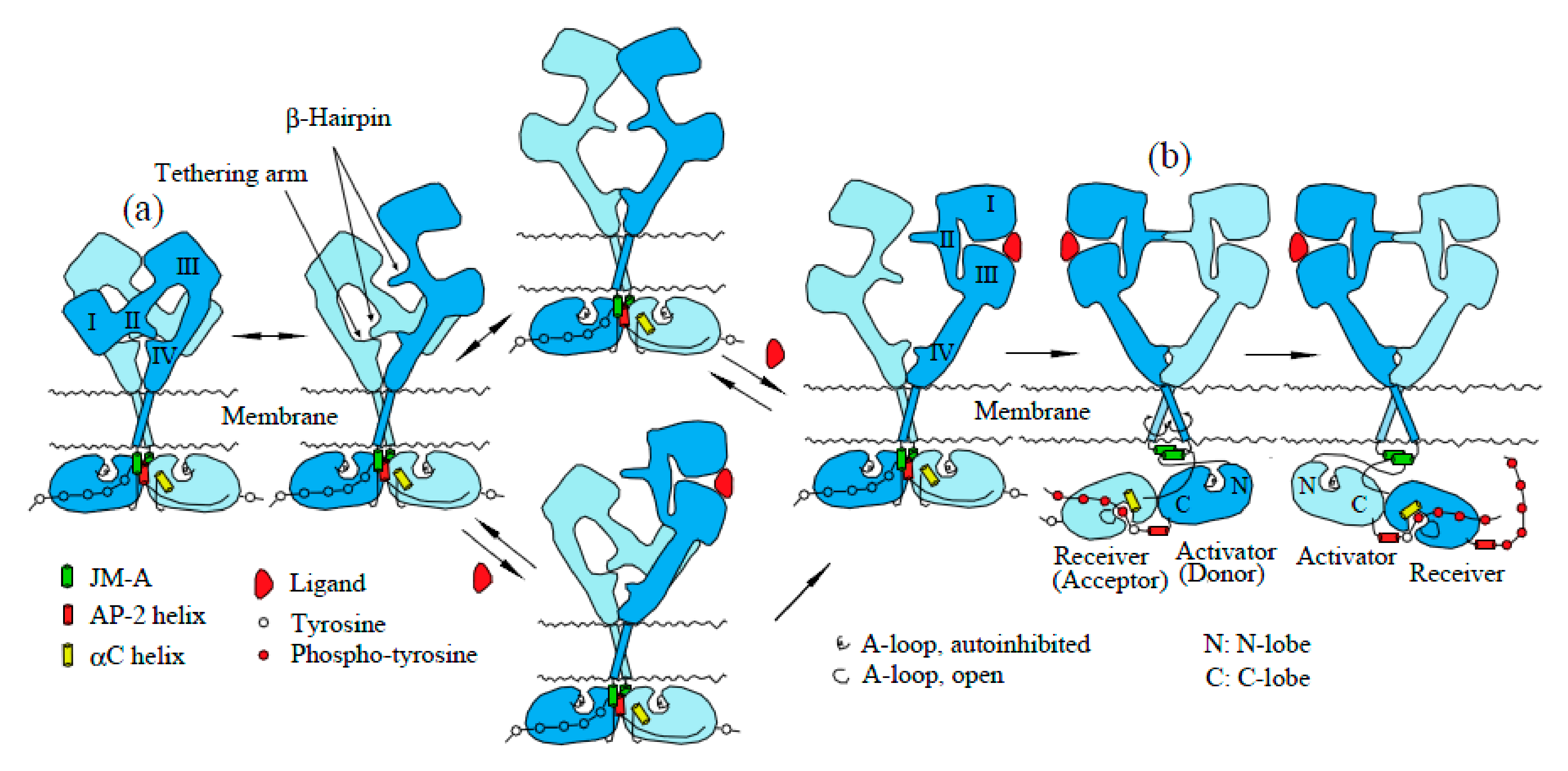
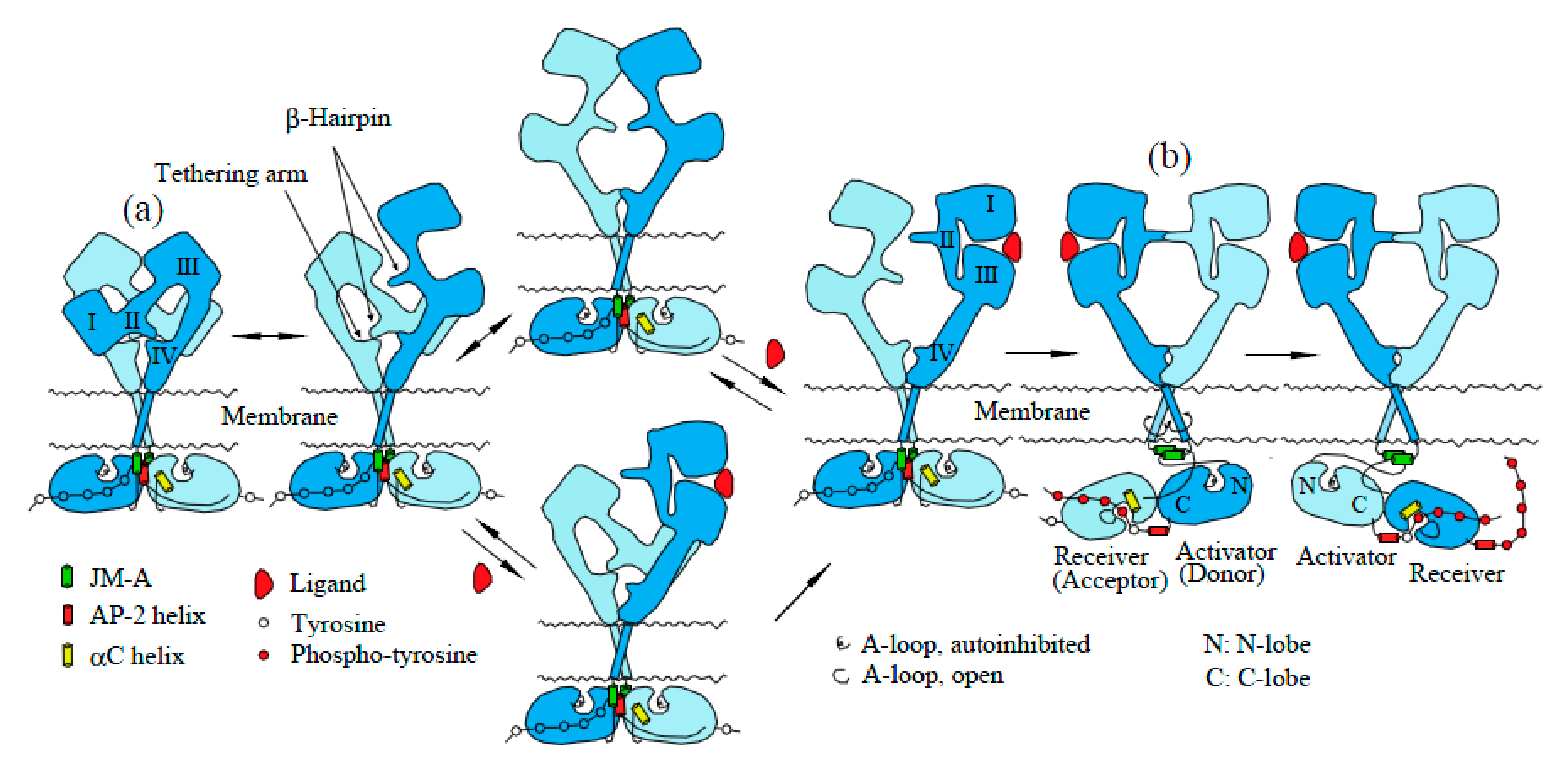
2.1. EGFR Has a Dimeric Structure
2.2. Structures of Inactive and Active EGFR Dimers
2.3. Negative Cooperativity in Ligand Binding to EGFR
2.4. Mechanism of Activation of EGFR Dimers by Ligand Binding: The “Rotation Model”
3. EGFR and Cancer
3.1. EGFR Overproduction
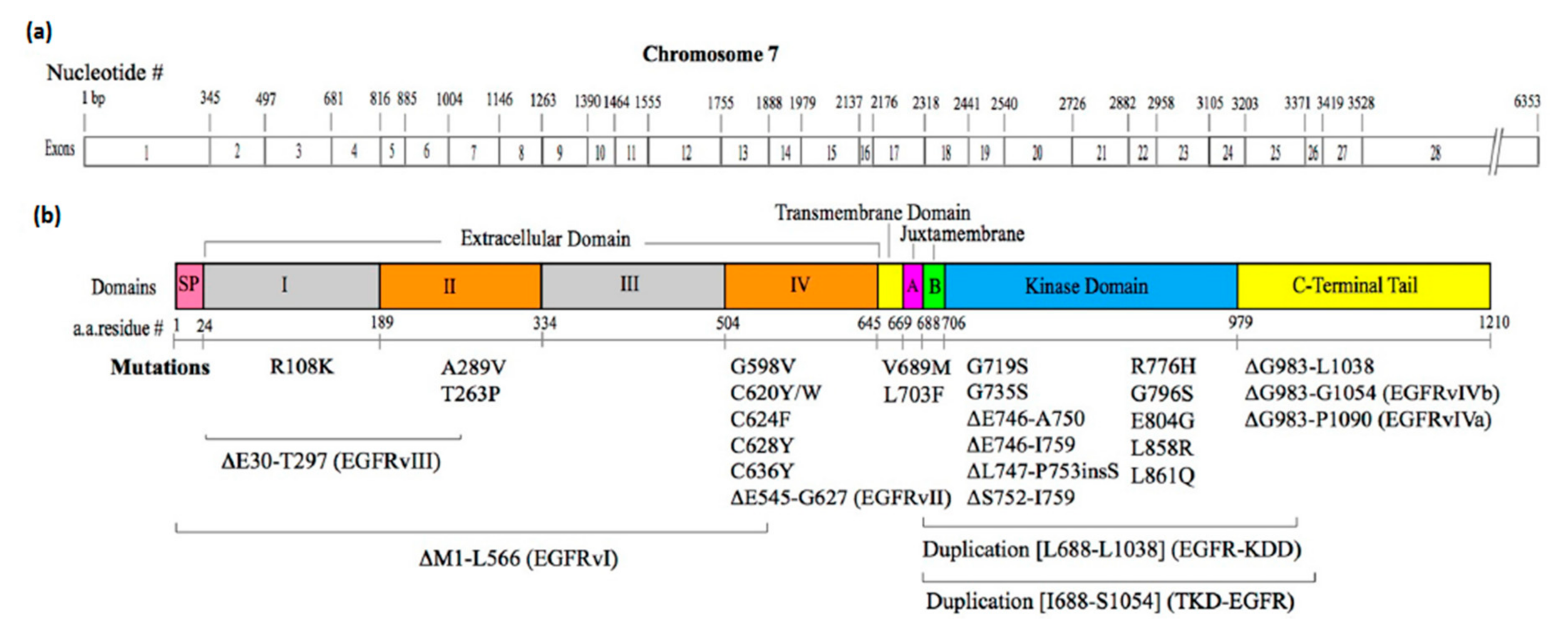
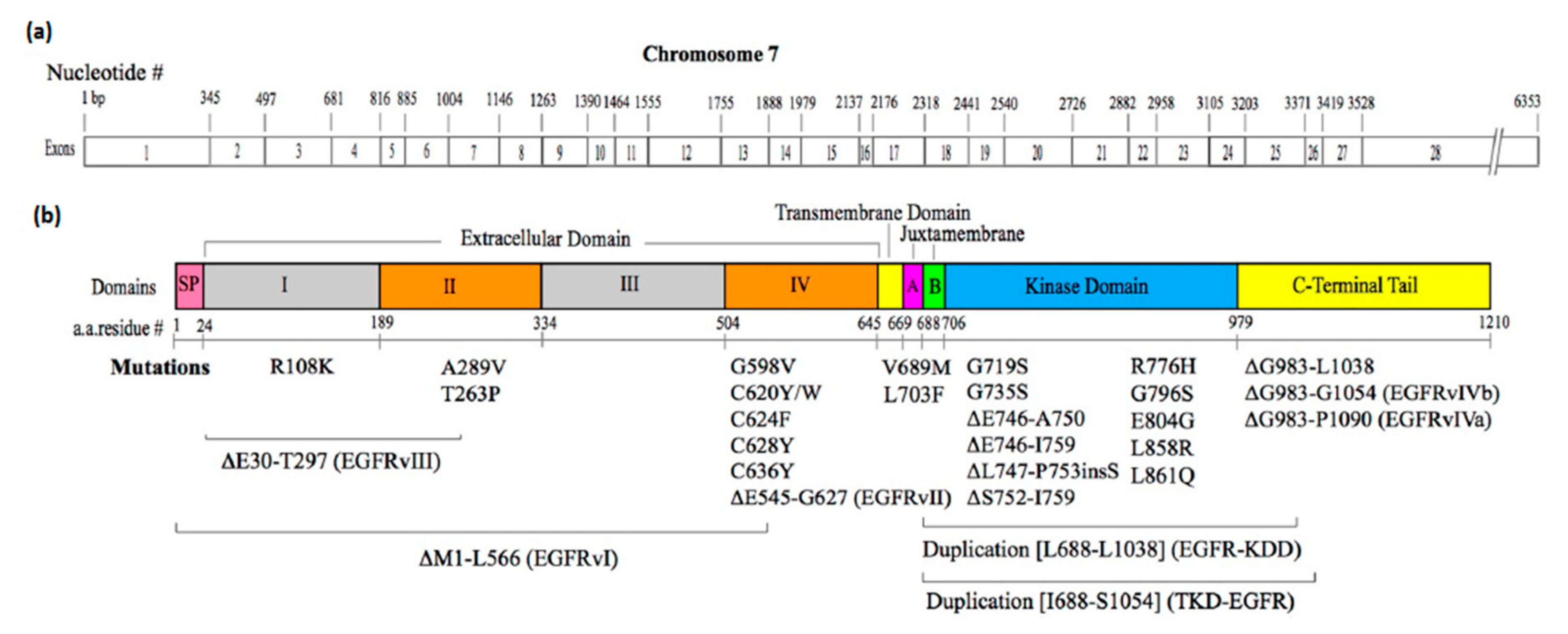
3.2. Mutations that Constitutively Activate EGFR
3.2.1. Mutations in the Extracellular Domain
3.2.2. Mutations in the Intracellular JM Region
3.2.3. Mutations in the Kinase Domain
3.2.4. Mutations in the C-Terminal Tail Region
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Rappoport, J.Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int. J. Biochem. Cell Biol. 2014, 51, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signaling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Burgess, A.W.; Cho, H.S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef]
- Ward, C.W.; Garrett, T.P.J. The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinform. 2001, 2, 4. [Google Scholar] [CrossRef]
- Ward, C.W.; Garrett, T.P.J.; McKern, N.M.; Lou, M.; Cosgrove, L.J.; Sparrow, L.G.; Frenkel, M.J.; Hoyne, P.A.; Elleman, T.C.; Adams, T.E.; et al. The three dimensional structure of the type I insulin-like growth factor receptor. Mol. Pathol. 2001, 54, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, R.A.; Chalkley, R.J.; Biarc, J.; Burlingame, A.L. Receptor tyrosine kinase signaling mechanisms: Devolving TrkA responses with phosphoproteomics. Adv. Biol. Regul. 2013, 53, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Taylor, P.; Jovceva, E.; St-Germain, J.R.; Jin, L.L.; Nikolic, A.; Gu, X.; Li, Z.H.; Trudel, S.; Moran, M.F. Tandem immunoprecipitation of phosphotyrosine-mass spectrometry (TIPY-MS) indicates C19ORF19 becomes tyrosine-phosphorylated and associated with activated epidermal growth factor receptor. J. Proteome Res. 2008, 7, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer 2002, 38, S3–S10. [Google Scholar] [CrossRef]
- Miloso, M.; Mazzotti, M.; Vass, W.C.; Beguinot, L. SHC and GRB-2 are constitutively activated by an epidermal growth factor receptor with a point mutation in the transmembrane domain. J. Biol. Chem. 1995, 270, 19557–19562. [Google Scholar] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Bessman, N.J.; Freed, D.M.; Lemmon, M.A. Putting together structures of epidermal growth factor receptors. Curr. Opin. Struct. Biol. 2014, 29, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Schlessinger, J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol. Cells 2010, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Pilch, P.F. Mechanism of epidermal growth factor receptor autophosphorylation and high-affinity binding. Proc. Natl. Acad. Sci. USA 1987, 84, 7832–7836. [Google Scholar] [CrossRef] [PubMed]
- Cochet, C.; Kashles, O.; Chambaz, E.M.; Borrello, I.; King, C.R.; Schlessinger, J. Demonstration of epidermal growth factor-induced receptor dimerization in living cells using a chemical covalent cross-linking agent. J. Biol. Chem. 1988, 263, 3290–3295. [Google Scholar] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Weiss, F.U.; Daub, H.; Ullrich, A. Novel mechanisms of RTK signal generation. Curr. Opin. Genet. Dev. 1997, 7, 80–86. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Maruyama, I.N. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model.” Bioessays 2015, 37, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Sudhaharan, T.; Koh, R.M.; Hwang, L.C.; Ahmed, S.; Maruyama, I.N.; Wohland, T. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 2007, 93, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef] [PubMed]
- Martin-Fernandez, M.; Clarke, D.T.; Tobin, M.J.; Jones, S.V.; Jones, G.R. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 2002, 82, 2415–2427. [Google Scholar] [CrossRef]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Yavas, S.; Machan, R.; Wohland, T. The epidermal growth factor receptor forms location-dependent complexes in resting cells. Biophys. J. 2016, 111, 2241–2254. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ahmed, S.; Wohland, T. EGFR activation monitored by SW-FCCS in live cells. Front. Biosci. (Elite Ed.) 2011, 3, 22–32. [Google Scholar] [PubMed]
- Hofman, E.G.; Bader, A.N.; Voortman, J.; van den Heuvel, D.J.; Sigismund, S.; Verkleij, A.J.; Gerritsen, H.C.; van Bergen en Henegouwen, P.M. Ligand-induced EGF receptor oligomerization is kinase-dependent and enhances internalization. J. Biol. Chem. 2010, 285, 39481–39489. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; En Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Teramura, Y.; Ichinose, J.; Takagi, H.; Nishida, K.; Yanagida, T.; Sako, Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J. 2006, 25, 4215–4222. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.H.; Maruyama, I.N. All EGF (ErbB) receptors have performed homo- and heterodimeric structures in living cells. J. Cell Sci. 2008, 121, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Ilagan, M.X.; Piwnica-Worms, D.; Pike, L.J. Luciferase fragment complementation imaging of conformational changes in the epidermal growth factor receptor. J. Biol. Chem. 2009, 284, 7474–7482. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Piwnica-Worms, D.; Pike, L.J. Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. USA 2012, 109, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Adak, S.; Landgraf, R.; Piwnica-Worms, D.; Pike, L.J. Dynamic analysis of the epidermal growth factor (EGF) receptor-ErbB2-ErbB3 protein network by luciferase fragment complementation imaging. J. Biol. Chem. 2013, 288, 30773–30784. [Google Scholar] [CrossRef] [PubMed]
- Knebel, A.; Rahmsdorf, H.J.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996, 15, 5314–5325. [Google Scholar] [PubMed]
- Yamashita, H.; Yano, Y.; Kawano, K.; Matsuzaki, K. Oligomerization-function relationship of EGFR on living cells detected by the coiled-coil labeling and FRET microscopy. Biochim. Biophys. Acta 2015, 1848, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Valley, C.C.; Arndt-Jovin, D.J.; Karedla, N.; Steinkamp, M.P.; Chizhik, A.I.; Hlavacek, W.S.; Wilson, B.S.; Lidke, K.A.; Lidke, D.S. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol. Biol. Cell 2015, 26, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. eLife 2016, 5, e14107. [Google Scholar] [CrossRef] [PubMed]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Moll. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Bouyain, S.; Longo, P.A.; Li, S.; Ferguson, K.M.; Leahy, D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 15024–15029. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; Mazzotti, M.; Sorkina, T.; Scotto, L.; Beguinot, L. Epidermal growth factor receptor interaction with clathrin adaptors is mediated by the Tyr974-containing internalization motif. J. Biol. Chem. 1996, 271, 13377–13384. [Google Scholar] [PubMed]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Schlessinger, J. Allosteric regulation of the epidermal growth factor receptor kinase. J. Cell Biol. 1986, 103, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.L.; Pike, L.J. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. USA 2008, 105, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Pike, L.J. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J. Biol. Chem. 2009, 284, 13570–13576. [Google Scholar] [CrossRef] [PubMed]
- Wofsy, C.; Goldstein, B.; Lund, K.; Wiley, H.S. Implications of epidermal growth factor (EGF) induced egf receptor aggregation. Biophys. J. 1992, 63, 98–110. [Google Scholar] [CrossRef]
- De Meyts, P.; Roth, J.; Neville, D.M., Jr.; Gavin, J.R., III; Lesniak, M.A. Insulin interactions with its receptors: Experimental evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 1973, 55, 154–161. [Google Scholar] [CrossRef]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 2010, 142, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cleveland, T.E., 4th; Bouyain, S.; Byrne, P.O.; Longo, P.A.; Leahy, D.J. A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. USA 2012, 109, 10861–10866. [Google Scholar] [CrossRef] [PubMed]
- Adak, S.; DeAndrade, D.; Pike, L.J. The tethering arm of the EGF receptor is required for negative cooperativity and signal transduction. J. Biol. Chem. 2011, 286, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Adak, S.; Yang, K.S.; Macdonald-Obermann, J.; Pike, L.J. The membrane-proximal intracellular domain of the epidermal growth factor receptor underlies negative cooperativity in ligand binding. J. Biol. Chem. 2011, 286, 45146–45155. [Google Scholar] [CrossRef] [PubMed]
- Shoyab, M.; De Larco, J.E.; Todaro, G.J. Biologically active phorbol esters specifically alter affinity of epidermal growth factor membrane receptors. Nature 1979, 279, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Magun, B.E.; Matrisian, L.M.; Bowden, G.T. Epidermal growth factor. Ability of tumor promoter to alter its degradation, receptor affinity and receptor number. J. Biol. Chem. 1980, 255, 6373–6381. [Google Scholar] [PubMed]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 1984, 311, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Downward, J.; Waterfield, M.D.; Parker, P.J. Autophosphorylation and protein kinase C phosphorylation of the epidermal growth factor receptor. Effect on tyrosine kinase activity and ligand binding affinity. J. Biol. Chem. 1985, 260, 14538–14546. [Google Scholar] [PubMed]
- Thiel, K.W.; Carpenter, G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc. Natl. Acad. Sci. USA 2007, 104, 19238–19243. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Merzlyakov, M.; Cohen, T.; Shai, Y.; Hristova, K. Energetics of ErbB1 transmembrane domain dimerization in lipid bilayers. Biophys. J. 2009, 96, 4622–4630. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Mineev, K.S.; Pavlov, K.V.; Akimov, S.A.; Kuznetsov, A.S.; Efremov, R.G.; Arseniev, A.S. Helix-helix interactions in membrane domains of bitopic proteins: Specificity and role of lipid environment. Biochim. Biophys. Acta 2017, 1859, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, I.N. Receptor Guanylyl Cyclases in Sensory Processing. Front. Endocrinol. 2017, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Lesovoy, D.M.; Pavlov, K.V.; Pustovalova, Y.E.; Bocharova, O.V.; Arseniev, A.S. Alternative packing of EGFR transmembrane domain suggests that protein-lipid interactions underlie signal conduction across membrane. Biochim. Biophys. Acta 2016, 1858, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.; Orchard, S.G.; Nice, E.C.; Posner, R.G.; Burgess, A.W. Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors 2008, 26, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Kozer, N.; Barua, D.; Henderson, C.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry 2014, 53, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.G.; Zhang, Y.; Ma, D.J.; Sarkaria, J.N.; White, F.M. MARQUIS: A multiplex method for absolute quantification of peptides and posttranslational modifications. Nat. Commun. 2015, 6, 5924. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Mill, C.; Lambert, S.; Buchman, J.; Wilson, T.R.; Hernandez-Gordillo, V.; Gallo, R.M.; Ades, L.M.; Settleman, J.; Riese, D.J., 2nd. EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 2012, 30, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Okada, S.; Ohshima, K.; Yamada, E.; Sato, M.; Uehara, Y.; Shimizu, H.; Pessin, J.E.; Mori, M. Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by betacellulin and EGF. Endocrinology 2004, 145, 4232–4243. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Pike, L.J. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem. 2014, 289, 26178–26188. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grovdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 2001, 8, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.W.; Tkach, V.; Pedersen, N.; Berezin, V.; Poulsen, H.S. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int. J. Cancer 2004, 108, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.W.; Pedersen, N.; Damstrup, L.; Villingshoj, M.; Sonder, S.U.; Rieneck, K.; Bovin, L.F.; Spang-Thomsen, M.; Poulsen, H.S. Analysis of the epidermal growth factor receptor specific transcriptome: Effect of receptor expression level and an activating mutation. J. Cell. Biochem. 2005, 96, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, S9–S15. [Google Scholar] [CrossRef]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR gene amplification in breast cancer: Correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Dobashi, Y.; Sakurai, H.; Nishikawa, K.; Hanawa, M.; Ooi, A. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas. An immunohistochemical and fluorescence in situ hybridization study. Cancer 2005, 103, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed]
- Furgason, J.M.; Li, W.; Milholland, B.; Cross, E.; Li, Y.; McPherson, C.M.; Warnick, R.E.; Rixe, O.; Stambrook, P.J.; Vijg, J.; et al. Whole genome sequencing of glioblastoma multiforme identifies multiple structural variations involved in EGFR activation. Mutagenesis 2014, 29, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.S.; Carrier, F.; Johnson, A.C.; Ogdon, S.E.; Fornace, A.J., Jr. Identification of an additional p53-responsive site in the human epidermal growth factor receptor gene promotor. Oncogene 1997, 15, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 1996, 16, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- McInerney, J.M.; Wilson, M.A.; Strand, K.J.; Chrysogelos, S.A. A strong intronic enhancer element of the EGFR gene is preferentially active in high EGFR expressing breast cancer cells. J. Cell. Biochem. 2001, 80, 538–549. [Google Scholar] [CrossRef]
- Chrysogelos, S.A. Chromatin structure of the EGFR gene suggests a role for intron 1 sequences in its regulation in breast cancer cells. Nucleic Acids Res. 1993, 21, 5736–5741. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, F.; Zanker, K.S.; Brandt, B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J. Biol. Chem. 1999, 274, 13176–13180. [Google Scholar] [CrossRef] [PubMed]
- Buerger, H.; Packeisen, J.; Boecker, A.; Tidow, N.; Kersting, C.; Bielawski, K.; Isola, J.; Yatabe, Y.; Nakachi, K.; Boecker, W.; et al. Allelic length of a CA dinucleotide repeat in the egfr gene correlates with the frequency of amplifications of this sequence—First results of an inter-ethnic breast cancer study. J. Pathol. 2004, 203, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Amador, M.L.; Oppenheimer, D.; Perea, S.; Maitra, A.; Cusatis, G.; Iacobuzio-Donahue, C.; Baker, S.D.; Ashfaq, R.; Takimoto, C.; Forastiere, A.; et al. An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res. 2004, 64, 9139–9143. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Hall, H.; DiLorenzo, T.P.; Steinberg, B.M. Elevation of the epidermal growth factor receptor and dependent signaling in human papillomavirus-infected laryngeal papillomas. Cancer Res. 1999, 59, 968–974. [Google Scholar] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Humphrey, P.A.; Gangarosa, L.M.; Wong, A.J.; Archer, G.E.; Lund-Johansen, M.; Bjerkvig, R.; Laerum, O.D.; Friedman, H.S.; Bigner, D.D. Deletion-mutant epidermal growth factor receptor in human gliomas: Effects of type II mutation on receptor function. Biochem. Biophys. Res. Commun. 1991, 178, 1413–1420. [Google Scholar] [CrossRef]
- Francis, J.M.; Zhang, C.Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar] [PubMed]
- Antonyak, M.A.; Moscatello, D.K.; Wong, A.J. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Everiss, K.D.; Wikstrand, C.J.; Batra, S.K.; Kung, H.J.; Bigner, D.D. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem. J. 1997, 324, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.D.; Huang, C.M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Greenall, S.A.; Donoghue, J.F.; Gottardo, N.G.; Johns, T.G.; Adams, T.E. Glioma-specific Domain IV EGFR cysteine mutations promote ligand-induced covalent receptor dimerization and display enhanced sensitivity to dacomitinib in vivo. Oncogene 2015, 34, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.Y.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef] [PubMed]
- Arjona, D.; Bello, M.J.; Alonso, M.E.; Aminoso, C.; Isla, A.; De Campos, J.M.; Sarasa, J.L.; Gutierrez, M.; Villalobo, A.; Rey, J.A. Molecular analysis of the EGFR gene in astrocytic gliomas: mRNA expression, quantitative-PCR analysis of non-homogeneous gene amplification and DNA sequence alterations. Neuropathol. Appl. Neurobiol. 2005, 31, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Idbaih, A.; Aimard, J.; Boisselier, B.; Marie, Y.; Paris, S.; Criniere, E.; Carvalho Silva, R.; Laigle-Donadey, F.; Rousseau, A.; Mokhtari, K.; et al. Epidermal growth factor receptor extracellular domain mutations in primary glioblastoma. Neuropathol. Appl. Neurobiol. 2009, 35, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Mendrola, J.M.; Lemmon, M.A. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene 2007, 26, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Greulich, H.; Jänne, P.A.; Sellers, W.R.; Meyerson, M.; Griffin, J.D. Epidermal growth factor—Independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces Gefitinib-sensitive cell cycle progression. Cancer Res. 2005, 65, 8968–8974. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Gullick, W.J.; Hill, M.E. Mutations of the epidermal growth factor receptor in non-small cell lung cancer—Search and destroy. Eur. J. Cancer 2006, 42, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, T.M.; Cable, L.; Alligood, K.; Rusnak, D.; Spehar, G.; Gallagher, K.T.; Woldu, E.; Carter, H.L.; Truesdale, A.T.; Shewchuk, L.; et al. Impact of common epidermal growth factor receptor and HER2 variants on receptor activity and inhibition by lapatinib. Cancer Res. 2008, 68, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.Y.; Chiu, C.H.; Li, L.H.; Hsiao, C.Y.; Tzen, C.Y.; Chang, K.T.; Chen, Y.M.; Perng, R.P.; Tsai, S.F.; Tsai, C.M. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non–small cell lung cancer. Clin. Cancer Res. 2005, 11, 3750–3757. [Google Scholar] [CrossRef] [PubMed]
- Van Noesel, J.; van der Ven, W.H.; van Os, T.A.; Kunst, P.W.; Weegenaar, J.; Reinten, R.J.; Kancha, R.K.; Duyster, J.; van Noesel, C.J. Activating germline R776H mutation in the epidermal growth factor receptor associated with lung cancer with squamous differentiation. J. Clin. Oncol. 2013, 31, e161–e164. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Kannan, N. Mechanistic insights into R776H mediated activation of epidermal growth factor receptor kinase. Biochemistry 2015, 54, 4216–4225. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Q.; Peng, Y.; Buckley, M.T.; Wei, J.; Chen, F.; Liebes, L.; Gerald, W.L.; Pincus, M.R.; Osman, I.; Lee, P. Epidermal growth factor receptor activation in prostate cancer by three novel missense mutations. Oncogene 2008, 27, 3201–3210. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. Uncommon GNAQ, MMP8, AKT3, EGFR, and PIK3R1 mutations in thyroid cancers. Endocr. Pathol. 2011, 22, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Schwentner, I.; Witsch-Baumgartner, M.; Sprinzl, G.M.; Krugmann, J.; Tzankov, A.; Jank, S.; Zwierzina, H.; Loeffler-Ragg, J. Identification of the rare EGFR mutation p.G796S as somatic and germline mutation in white patients with squamous cell carcinoma of the head and neck. Head Neck 2008, 30, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Ciesielski, M.J.; Castiglia, G.J. Tandem duplication of the epidermal growth factor receptor tyrosine kinase and calcium internalization domains in A-172 glioma cells. Oncogene 1998, 16, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Ozer, B.H.; Wiepz, G.J.; Bertics, P.J. Activity and cellular localization of an oncogenic glioblastoma multiforme-associated EGF receptor mutant possessing a duplicated kinase domain. Oncogene 2010, 29, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.N.; Sheehan, J.H.; Shaver, T.M.; Bailey, M.; Lipson, D.; Chandramohan, R.; Red Brewer, M.; York, S.J.; Kris, M.G.; Pietenpol, J.A.; et al. EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer That Is Clinically Responsive to Afatinib. Cancer Discov. 2015, 5, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust detection of EGFR copy number changes and EGFR variant III: Technical aspects and relevance for glioma diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 2001, 8, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Huang, P.H.; Zwang, Y.; White, F.M.; Yarden, Y. EGFRvIV: A previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 2010, 29, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Pastorino, S.; Zeng, Q.; Xu, X.; Johnson, W.; Vandenberg, S.; Verhaak, R.; Cherniack, A.D.; Watanabe, H.; Dutt, A.; et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Res. 2011, 71, 7587–7596. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Francis, J.M.; Park, W.Y.; Park, J.O.; Cho, J. Constitutive asymmetric dimerization drives oncogenic activation of epidermal growth factor receptor carboxyl-terminal deletion mutants. Oncotarget 2015, 6, 8839–8850. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purba, E.R.; Saita, E.-i.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. https://doi.org/10.3390/cells6020013
Purba ER, Saita E-i, Maruyama IN. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells. 2017; 6(2):13. https://doi.org/10.3390/cells6020013
Chicago/Turabian StylePurba, Endang R., Ei-ichiro Saita, and Ichiro N. Maruyama. 2017. "Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”" Cells 6, no. 2: 13. https://doi.org/10.3390/cells6020013
APA StylePurba, E. R., Saita, E.-i., & Maruyama, I. N. (2017). Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells, 6(2), 13. https://doi.org/10.3390/cells6020013