
Autophagy-Related Deubiquitinating Enzymes Involved in Health and Disease


Abstract
:1. Introduction
1.1. Autophagy
1.2. Deubiquitinating Enzymes
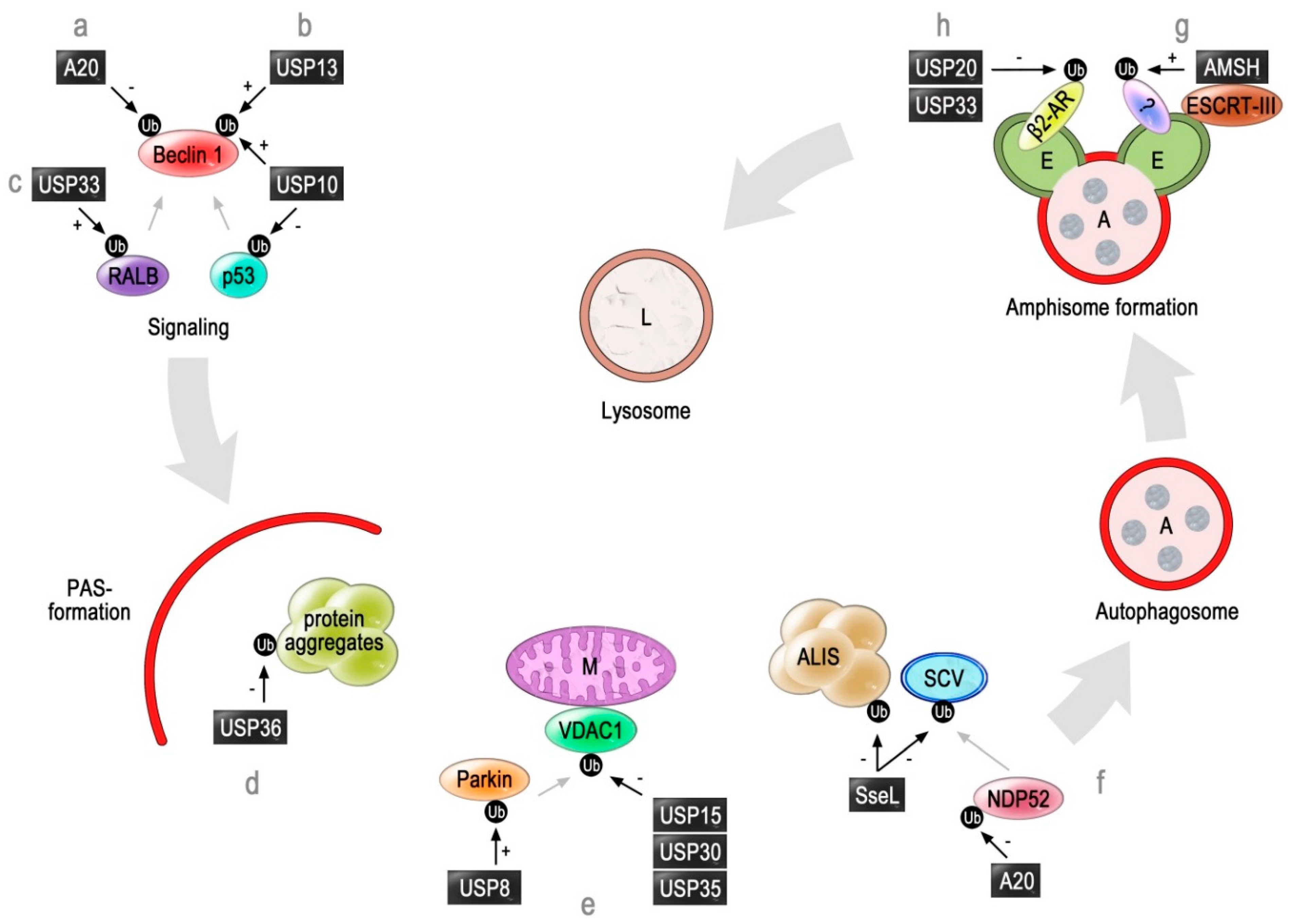
2. Deubiquitinating Enzymes Involved in Autophagy
2.1. A20
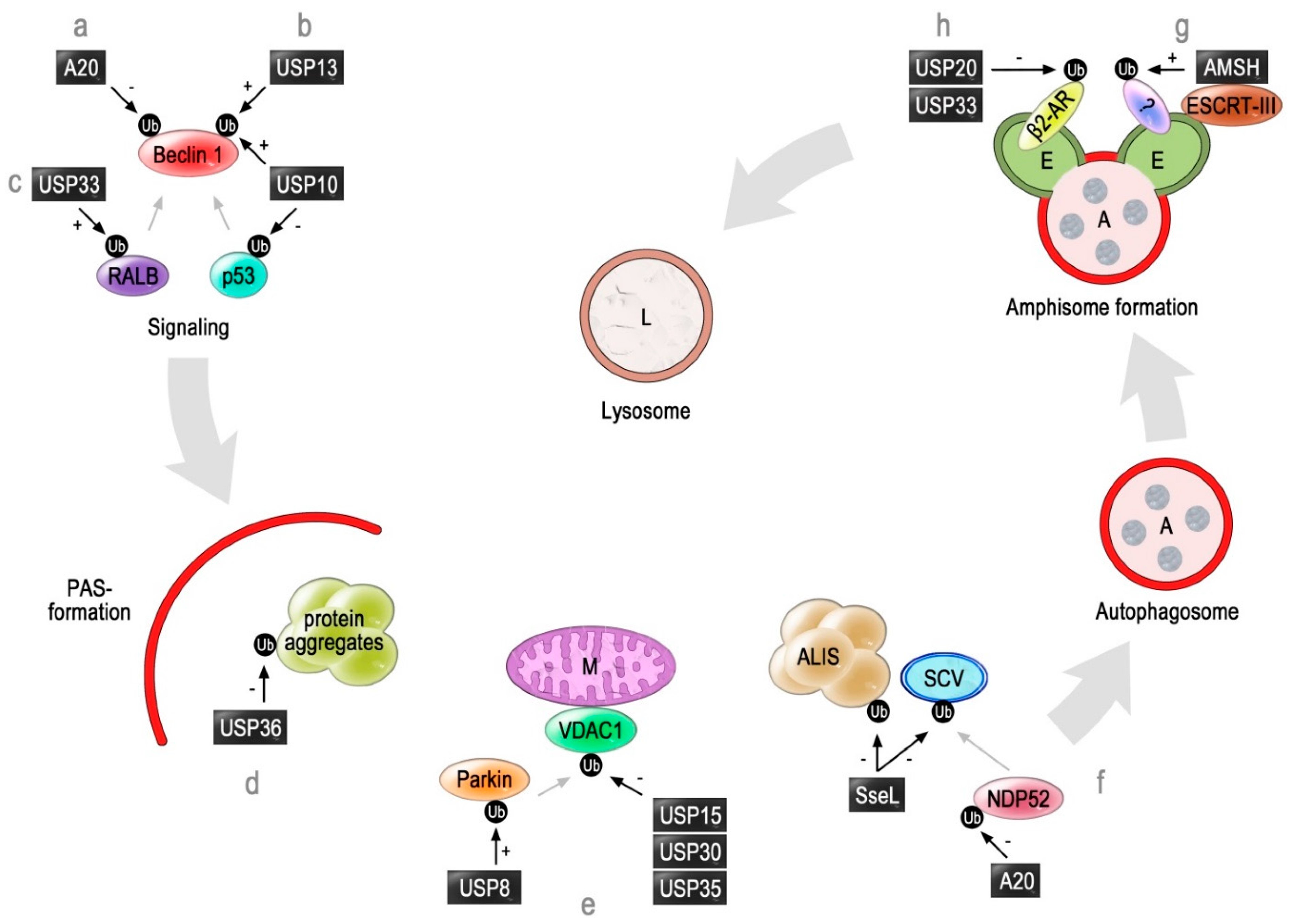
2.2. USP10 and USP13
2.3. USP33 and USP20
2.4. AMSH
2.5. USP8, USP15, USP30 and USP35
2.6. USP36
2.7. SseL
3. Conclusions

{kind=link}
| Deubiquitinase | DUB-type | Localization | Involvement in Autophagy 1 | Autophagy-related Target 1 | Effect on Autophagy | Disease Category 1,2 | Inhibitor 3 | References |
|---|---|---|---|---|---|---|---|---|
| A20 (TNFAIP3) | OTU | cytosol , lysosome |
|
| - - | Inflammation; Cancer? | [57,58,59,60,61,63,65,76] | |
| AMSH | JAMM | endosome |
|
| + | Neuro-degeneration | [109,110,111] | |
| SseL | bacterial DUB | cytosol |
|
| - | Bacterial infection | [173] | |
| UCH-L1 | UCH | ER, cytosol, nucleus |
|
| - | Neuro-degeneration | LDN91946; Isa- tin O-acyloxime | [53,54,55] |
| USP8 (UBPY) | USP | cytosol, endosome |
|
| + | Neuro-degeneration? | HBX 90,397; 9-E | [131,136,137,138] |
| USP10 | USP | cytosol, nucleus |
|
| + - | Cancer | Spautin 1 | [79,82,83,89,91] |
| USP13 | USP | cytosol, nucleus |
|
| + | Cancer | Spautin 1 | [79,82,83] |
| USP15 | USP | mitochondrion |
|
| - | Neuro-degeneration | [139] | |
| USP20 | USP | cytosol, cytoskeleton |
|
| - | Cancer | [102,105] | |
| USP22 | USP | cytosol, nucleus |
|
| + | Cancer | [51] | |
| USP30 | USP | mitochondrion |
|
| - | Neuro-degeneration | 15-oxospira-milactone(S3) | [140,141,143,145] |
| USP33 | USP | cytosol, cytoskeleton, Golgi apparatus |
|
| + - | Cancer | [94,95,96,97,102] | |
| USP35 | USP | mitochondrion |
|
| - | Neuro-degeneration | [141] | |
| USP36 | USP | cytosol, nucleus |
|
| - | Cancer, Neuro-degeneration | [158,161,162] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Stenmark, H. Endocytosis and signaling. Curr. Opin. Cell. Biol. 2011, 23, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Girardin, S.E. Emerging themes in bacterial autophagy. Curr. Opin. Microbiol. 2015, 23, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell. Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell. Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell. Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Eskelinen, E.L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. How do ESCRT proteins control autophagy? J. Cell. Sci. 2009, 122, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Kwon, Y.T.; Kim, B.Y. Amino-terminal arginylation as a degradation signal for selective autophagy. BMB Rep. 2015, 48, 487–488. [Google Scholar] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell. Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Fujioka, Y. Atg1 family kinases in autophagy initiation. Cell. Mol. Life Sci. 2015, 72, 3083–3096. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, H.; Stenmark, H.; Platta, H.W. Ubiquitination and phosphorylation of Beclin 1 and its binding partners: Tuning class III phosphatidylinositol 3-kinase activity and tumor suppression. FEBS Lett. 2012, 586, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell. Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell. Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Beaumelle, B.; Vergne, I. Autophagy in Mycobacterium tuberculosis and HIV infections. Front. Cell. Infect. Microbiol. 2015, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Yuan, J.; Jung, Y.K. Autophagy in Neurodegenerative Diseases: From Mechanism to Therapeutic Approach. Mol. Cells 2015, 38, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Invest. 2015, 125, 47–54. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-mediated tumor promotion. Cell. 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Reidick, C.; El Magraoui, F.; Meyer, H.E.; Stenmark, H.; Platta, H.W. Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO. Cancers 2015, 7, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Qi, J.; Ronai, Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 2013, 38, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.D.; Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 2003, 278, 35857–35860. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Larsen, C.N.; Cook, W.J.; Wilkinson, K.D.; Hill, C.P. Crystal structure of a deubiquitinating enzyme (human UCH-L3) at 1.8 A resolution. EMBO J. 1997, 16, 3787–3796. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002, 111, 1041–1054. [Google Scholar] [CrossRef]
- Komander, D.; Barford, D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem. J. 2008, 409, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Messick, T.E.; Russell, N.S.; Iwata, A.J.; Sarachan, K.L.; Shiekhattar, R.; Shanks, J.R.; Reyes-Turcu, F.E.; Wilkinson, K.D.; Marmorstein, R. Structural basis for ubiquitin recognition by the Otu1 ovarian tumor domain protein. J. Biol. Chem. 2008, 283, 11038–11049. [Google Scholar] [CrossRef] [PubMed]
- Nanao, M.H.; Tcherniuk, S.O.; Chroboczek, J.; Dideberg, O.; Dessen, A.; Balakirev, M.Y. Crystal structure of human otubain 2. EMBO Rep. 2004, 5, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Enesa, K.; Evans, P. The biology of A20-like molecules. Adv. Exp. Med. Biol. 2014, 809, 33–48. [Google Scholar] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell. Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbé, S. Deubiquitylases From Genes to Organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta. 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Debelyy, M.O.; Platta, H.W.; Saffian, D.; Hensel, A.; Thoms, S.; Meyer, H.E.; Warscheid, B.; Girzalsky, W.; Erdmann, R. Ubp15p, a ubiquitin hydrolase associated with the peroxisomal export machinery. J. Biol. Chem. 2011, 286, 28223–28234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Satoh, A.; Warren, G.; Meyer, H.H. VCIP135 acts as a deubiquitinating enzyme during p97-p47-mediated reassembly of mitotic Golgi fragments. J. Cell. Biol. 2004, 164, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Isono, E.; Katsiarimpa, A.; Müller, I.K.; Anzenberger, F.; Stierhof, Y.D.; Geldner, N.; Chory, J.; Schwechheimer, C. The deubiquitinating enzyme AMSH3 is required for intracellular trafficking and vacuole biogenesis in Arabidopsis thaliana. Plant. Cell. 2010, 22, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Seiberlich, V.; Borchert, J.; Zhukareva, V.; Richter-Landsberg, C. Inhibition of protein deubiquitination by PR-619 activates the autophagic pathway in OLN-t40 oligodendroglial cells. Cell. Biochem. Biophys. 2013, 67, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.I.; Coughlin, K.; Scotti, A.; Iizuka, Y.; Anchoori, R.; Roden, R.B.; Marastoni, M.; Bazzaro, M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015, 6, 4159–4170. [Google Scholar] [CrossRef] [PubMed]
- Drießen, S.; Berleth, N.; Friesen, O.; Löffler, A.S.; Böhler, P.; Hieke, N.; Stuhldreier, F.; Peter, C.; Schink, K.O.; Schultz, S.W.; et al. Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy 2015, 11, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.X.; Ning, Z.; Gao, W.; Ling, J.; Wang, A.M.; Luo, H.F.; Liang, Y.; Yan, Q.; Wang, Z.Y. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014, 32, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Pukaß, K.; Richter-Landsberg, C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents α-synuclein aggregate formation by activating the autophagic pathway: Implications for multiple system atrophy. Front. Cell. Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, L.; Niu, T.; Shao, L.; Li, H.; Wu, W.; Wang, W.; Lv, L.; Qin, Q.; Wang, F.; et al. Ubiquitin carboxyl terminal hydrolyase L1-suppressed autophagic degradation of p21WAF1/Cip1 as a novel feedback mechanism in the control of cardiac fibroblast proliferation. PLoS ONE 2014, 9, e94658. [Google Scholar] [CrossRef] [PubMed]
- Mermerian, A.H.; Case, A.; Stein, R.L.; Cuny, G.D. Structure-activity relationship, kinetic mechanism, and selectivity for a new class of ubiquitin C-terminal hydrolase-L1 (UCH-L1) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3729–3732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lashuel, H.A.; Choi, S.; Xing, X.; Case, A.; Ni, J.; Yeh, L.A.; Cuny, G.D.; Stein, R.L.; Lansbury, P.T.J. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 2003, 10, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Pujari, R.; Hunte, R.; Khan, W.N.; Shembade, N. A20-mediated negative regulation of canonical NF-κB signaling pathway. Immunol. Res. 2013, 57, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Troppan, K.; Hofer, S.; Wenzl, K.; Lassnig, M.; Pursche, B.; Steinbauer, E.; Wiltgen, M.; Zulus, B.; Renner, W.; Beham-Schmid, C.; et al. Frequent down regulation of the tumor suppressor gene a20 in multiple myeloma. PLoS ONE 2015, 10, e0123922. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Kim, D.H.; Lee, K.J.; Kim, C.S.; Song, S.B.; Cai, D.Q.; Kim, S.K. Autophagy contributes to apoptosis in A20 and EL4 lymphoma cells treated with fluvastatin. Cancer Cell. Int. 2013, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.E.; Turer, E.E.; Taylor, K.E.; Fang, C.J.; Advincula, R.; Oshima, S.; Barrera, J.; Huang, E.J.; Hou, B.; Malynn, B.A.; et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat. Immunol. 2011, 12, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Boonyasrisawat, W.; Eberle, D.; Bacci, S.; Zhang, Y.Y.; Nolan, D.; Gervino, E.V.; Johnstone, M.T.; Trischitta, V.; Shoelson, S.E.; Doria, A. Tag polymorphisms at the A20 (TNFAIP3) locus are associated with lower gene expression and increased risk of coronary artery disease in type 2 diabetes. Diabetes 2007, 56, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Beyaert, R.; van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 238, 33175–33182. [Google Scholar] [CrossRef] [PubMed]
- Erpapazoglou, Z.; Walker, O.; Haguenauer-Tsapis, R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells 2014, 3, 1027–1088. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy 2010, 6, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, S.; Du, Y.; Zhao, Z.; Shi, L.; Sun, L.; Huang, G.; Ye, B.; Li, C.; Dai, Z.; et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J. 2013, 32, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell. Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Inomata, M.; Niida, S.; Shibata, K.; Into, T. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell. Mol. Life Sci. 2012, 69, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Von Muhlinen, N.; Thurston, T.; Ryzhakov, G.; Bloor, S.; Randow, F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010, 6, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Inoue, M.; Danzaki, K.; Hammer, G.; He, Y.W.; Shinohara, M.L. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat. Commun. 2015, 22, 5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell. Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, S.B.; Pedersen, N.M.; Liestøl, K.; Stenmark, H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp. Cell. Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.J.; Valdes, Y.R.; Peart, T.M.; Fazio, E.N.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis 2014, 35, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell. Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell. Biol. 2013, 15, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell. Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Jochemsen, A.G.; Shiloh, Y. USP10: Friend and foe. Cell. 2010, 140, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, T.; Xu, D.; Wang, H.; Cai, Y.; Jin, T.; Liu, M.; Jin, M.; Wu, K.; Yuan, J. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev. 2015, 29, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wen, P.; Kong, R.; Cheng, H.; Zhang, B.; Quan, C.; Bian, Z.; Chen, M.; Zhang, Z.; Chen, X.; et al. USP33 mediates Slit-Robo signaling in inhibiting colorectal cancer cell migration. Int. J. Cancer 2015, 136, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.; Kong, R.; Liu, J.; Zhu, L.; Chen, X.; Li, X.; Nie, Y.; Wu, K.; Wu, J.Y. USP33, a new player in lung cancer, mediates Slit-Robo signaling. Protein Cell. 2014, 5, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; D’Angiolella, V.; Seeley, E.S.; Kim, S.; Kobayashi, T.; Fu, W.; Campos, E.I.; Pagano, M.; Dynlacht, B.D. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 2013, 495, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Simicek, M.; Lievens, S.; Laga, M.; Guzenko, D.; Aushev, V.N.; Kalev, P.; Baietti, M.F.; Strelkov, S.V.; Gevaert, K.; Tavernier, J.; et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell. Biol. 2013, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, R.; Horiuchi, H. Ral GTPases: Crucial mediators of exocytosis and tumourigenesis. J. Biochem. 2015, 157, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M.J.; et al. RalB GTPase-mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Komici, K.; Corbi, G.; Pagano, G.; Furgi, G.; Rengo, C.; Femminella, G.D.; Leosco, D.; Bonaduce, D. β-adrenergic receptor responsiveness in aging heart and clinical implications. Front. Physiol. 2014, 4, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 2008, 283, 22166–22176. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Jean-Charles, P.Y.; Shenoy, S.K. Phosphorylation of the Deubiquitinase USP20 by Protein Kinase A Regulates Post-endocytic Trafficking of β2 Adrenergic Receptors to Autophagosomes during Physiological Stress. J. Biol. Chem. 2015, 290, 8888–8903. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kaneko, K.; Asao, H.; Kasai, H.; Endo, Y.; Fujita, T.; Takeshita, T.; Sugamura, K. Possible involvement of a novel STAM-associated molecule “AMSH” in intracellular signal transduction mediated by cytokines. J. Biol. Chem. 1999, 274, 19129–19135. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Clague, M.J.; Urbé, S. AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell. Biol. 2004, 166, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Kyuuma, M.; Kikuchi, K.; Kojima, K.; Sugawara, Y.; Sato, M.; Mano, N.; Goto, J.; Takeshita, T.; Yamamoto, A.; Sugamura, K.; et al. AMSH, an ESCRT-III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell. Struct. Funct. 2007, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Agromayor, M.; Martin-Serrano, J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J. Biol. Chem. 2006, 281, 23083–23091. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Owada, Y.; Yamada, M.; Miura, S.; Murata, K.; Asao, H.; Kondo, H.; Sugamura, K. Loss of neurons in the hippocampus and cerebral cortex of AMSH-deficient mice. Mol. Cell. Biol. 2001, 21, 8626–8637. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tamai, K.; Watanabe, M.; Kyuuma, M.; Ono, M.; Sugamura, K.; Tanaka, N. AMSH is required to degrade ubiquitinated proteins in the central nervous system. Biochem. Biophys. Res. Commun. 2011, 408, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell. Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Liu, L.; Gao, F.B. Autophagy defects contribute to neurodegeneration induced by dysfunctional ESCRT-III. Autophagy 2009, 5, 1070–1072. [Google Scholar] [CrossRef] [PubMed]
- Katsiarimpa, A.; Kalinowska, K.; Anzenberger, F.; Weis, C.; Ostertag, M.; Tsutsumi, C.; Schwechheimer, C.; Brunner, F.; Hückelhoven, R.; Isono, E. The deubiquitinating enzyme AMSH1 and the ESCRT-III subunit VPS2.1 are required for autophagic degradation in Arabidopsis. Plant. Cell. 2013, 25, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Katsiarimpa, A.; Anzenberger, F.; Schlager, N.; Neubert, S.; Hauser, M.T.; Schwechheimer, C.; Isono, E. The Arabidopsis deubiquitinating enzyme AMSH3 interacts with ESCRT-III subunits and regulates their localization. Plant. Cell. 2011, 23, 3026–3040. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Müller-Rischart, A.K.; Pilsl, A.; Beaudette, P.; Patra, M.; Hadian, K.; Funke, M.; Peis, R.; Deinlein, A.; Schweimer, C.; Kuhn, P.H.; et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell. 2013, 49, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell. Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb. Perspect. Biol. 2012, 4, a011338. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell. Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W.N. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Vincow, E.S.; Merrihew, G.; Thomas, R.E.; Shulman, N.J.; Beyer, R.P.; MacCoss, M.J.; Pallanck, L.J. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6400–6405. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell. Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Che, L.; Ren, J.; Pandita, R.K.; Lu, J.; Li, K.; Pandita, T.K.; Du, C. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc. Natl. Acad. Sci. USA 2015, 112, E1210–E1219. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shi, Y.; Zhao, Y. USP8 mutation in Cushing’s disease. Oncotarget. 2015, 6, 18240–18241. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, M.J.; Nicholson, B.; Kessler, B.M. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert. Rev. Mol. Med. 2011, 13, e35. [Google Scholar] [CrossRef] [PubMed]
- Guedat, P.; Boissy, G.; Borg-Capra, C.; Colland, F.; Daviet, L.; Formstecher, E.; Jacq, X.; Rain, J.-C.; Delansorne, R.; Vallese, S.; et al. Novel Cysteine Protease Inhibitors and Their Therapeutic Applications. WO2007017758, 15 February 2007. [Google Scholar]
- Byun, S.; Lee, S.Y.; Lee, J.; Jeong, C.H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.H.; Lee, H.J.; et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Haddad, D.; Wauters, F.; van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; de Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three “P”s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell. Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Thobois, S. USP30: A new promising target for Parkinson’s disease? Mov. Disord. 2015, 30, 340. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Yan, C.; Wu, H.; Du, L.; Wang, Y.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell. Res. 2014, 24, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Hirose, S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol. Biol. Cell. 2008, 19, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M. Mitofusins: Ubiquitylation promotes fusion. Cell. Res. 24, 387–388. [CrossRef] [PubMed]
- Jaeger, P.A.; Wyss-Coray, T. Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 2010, 67, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.S.; Rebeck, G.W.; Moussa, C.E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Aβ in AD models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, P.A.; Pickford, F.; Sun, C.H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS ONE 2010, 5, e11102. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Invest. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Gao, F.B. Regulation of Aβ pathology by beclin 1: A protective role for autophagy? J. Clin. Invest. 2008, 118, 2015–2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.W.; Bandyopadhyay, U.; Jiang, Y.; et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011, 7, 788–789. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Kontogiannea, M.; Thorarinsdottir, T.; Fallon, L.; Williams, A.J.; Djarmati, A.; Fantaneanu, T.; Paulson, H.L.; Fon, E.A. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 2011, 20, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell. Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Taillebourg, E.; Gregoire, I.; Viargues, P.; Jacomin, A.C.; Thevenon, D.; Faure, M.; Fauvarque, M.O. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy 2012, 8, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Casey, S.C.; Felsher, D.W. Inactivation of MYC reverses tumorigenesis. J. Intern. Med. 2014, 276, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhangm, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell. Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yoo, K.J.; Kang, I.; Chung, H.M.; Baek, K.H. A novel cysteine protease HeLa DUB-1 responsible for cleaving the ubiquitin in human ovarian cancer cells. Int. J. Oncol. 2004, 25, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; D’Orazio, M.; Cecconi, F. AMBRA1 and BECLIN 1 interplay in the crosstalk between autophagy and cell proliferation. Cell. Cycle 2015, 14, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; de Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell. Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell. Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Coombes, B.K.; Lowden, M.J.; Bishop, J.L.; Wickham, M.E.; Brown, N.F.; Duong, N.; Osborne, S.; Gal-Mor, O.; Finlay, B.B. SseL is a salmonella-specific translocated effector integrated into the SsrB-controlled salmonella pathogenicity island 2 type III secretion system. Infect. Immun. 2007, 75, 574–580. [Google Scholar] [CrossRef] [PubMed]
- De Jong, H.K.; Parry, C.M.; van der Poll, T.; Wiersinga, W.J. Host-pathogen interaction in invasive Salmonellosis. PLoS Pathog. 2012, 8, e1002933. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. Autophagy in immunity against intracellular bacteria. Curr. Top. Microbiol. Immunol. 2009, 335, 189–215. [Google Scholar] [PubMed]
- Huang, J.; Brumell, J.H. Bacteria-autophagy interplay: A battle for survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Brumell, J.H. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2006, 2, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Szeto, J.; Kaniuk, N.A.; Canadien, V.; Nisman, R.; Mizushima, N.; Yoshimori, T.; Bazett-Jones, D.P.; Brumell, J.H. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006, 2, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.; Figueira, R.; Holden, D.W. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Mesquita, F.S.; Holden, D.W. The DUB-ious lack of ALIS in Salmonella infection: A Salmonella deubiquitinase regulates the autophagy of protein aggregates. Autophagy 2012, 8, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-κB, suppresses IκBα ubiquitination and modulates innate immune responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Holden, D.W.; Rolhion, N. Lack of effect of the Salmonella deubiquitinase SseL on the NF-κB pathway. PLoS ONE 2013, 8, e53064. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magraoui, F.E.; Reidick, C.; Meyer, H.E.; Platta, H.W. Autophagy-Related Deubiquitinating Enzymes Involved in Health and Disease. Cells 2015, 4, 596-621. https://doi.org/10.3390/cells4040596
Magraoui FE, Reidick C, Meyer HE, Platta HW. Autophagy-Related Deubiquitinating Enzymes Involved in Health and Disease. Cells. 2015; 4(4):596-621. https://doi.org/10.3390/cells4040596
Chicago/Turabian StyleMagraoui, Fouzi El, Christina Reidick, Hemut E. Meyer, and Harald W. Platta. 2015. "Autophagy-Related Deubiquitinating Enzymes Involved in Health and Disease" Cells 4, no. 4: 596-621. https://doi.org/10.3390/cells4040596
APA StyleMagraoui, F. E., Reidick, C., Meyer, H. E., & Platta, H. W. (2015). Autophagy-Related Deubiquitinating Enzymes Involved in Health and Disease. Cells, 4(4), 596-621. https://doi.org/10.3390/cells4040596