
Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS


Abstract
:1. Autophagy: Main Features and Links with the Ubiquitin-Proteasome System
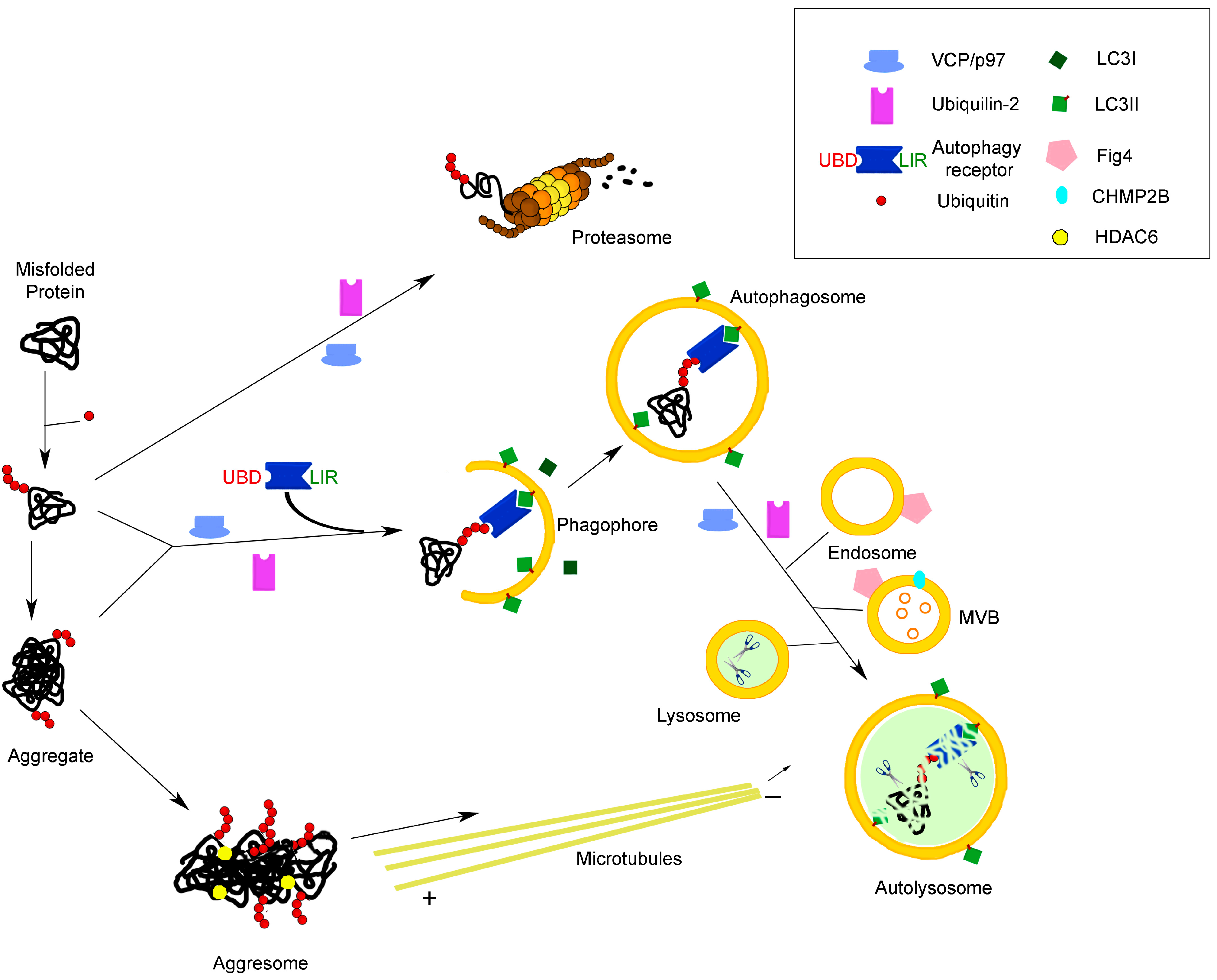
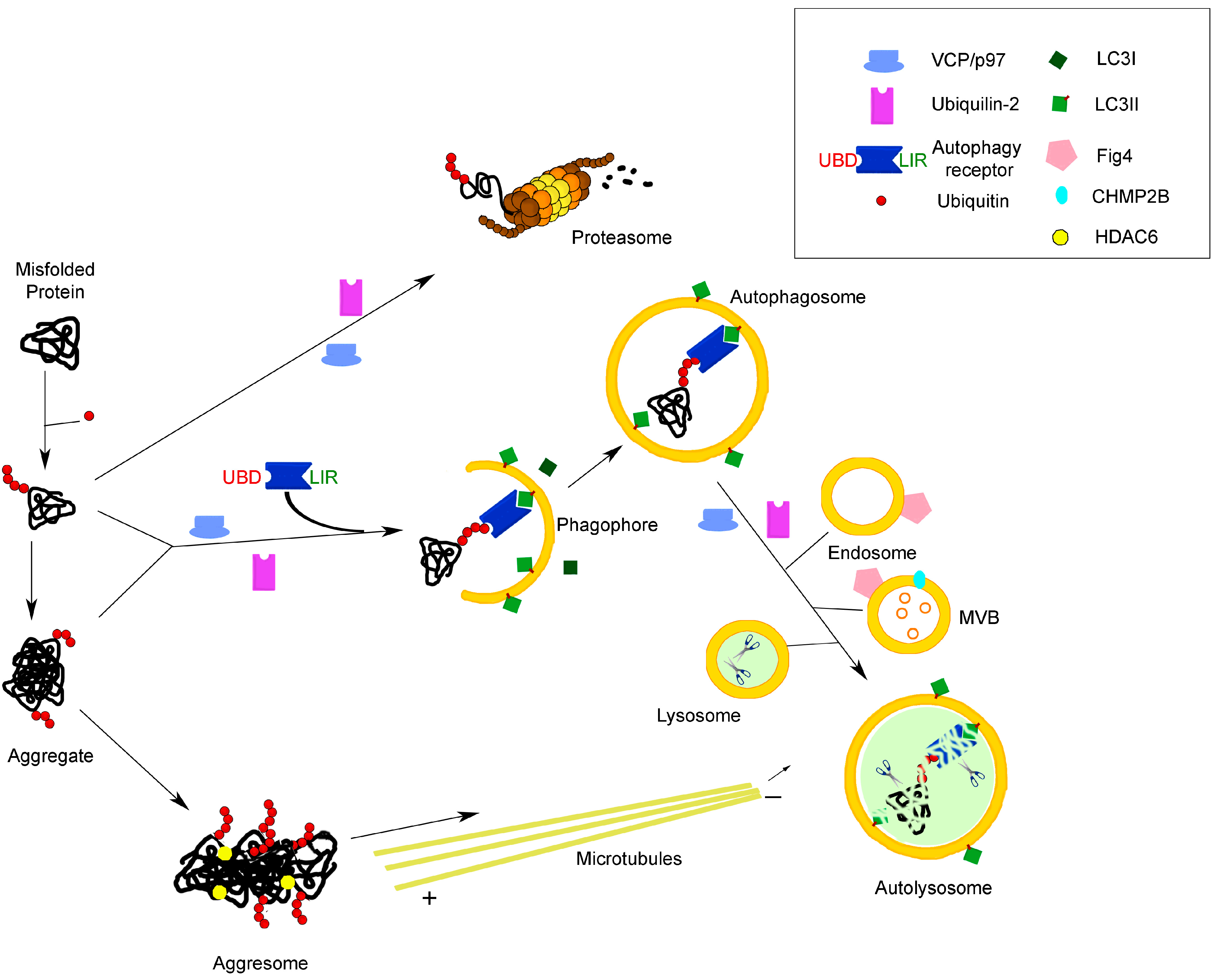
2. Autophagy and Neurodegeneration
3. Autophagy in the Pathogenesis of ALS

{kind=link}
{kind=link}
| Mutant Molecule | Gene Locus | Inheritance | Predominant Phenotype | Autophagy Defect | Estimated % | Reference |
|---|---|---|---|---|---|---|
| SQSTH1/p62 (sequestosome) | 5q35 | Dominant | ALS/ALS + FTLD | Substrate recognition Selective autophagy | Unknown | [86] Reviewed in [32] |
| Optineurin | 10p15-p14 | Dominant: missense mutations; Recessive: Exon 5 deletions and nonsense mutations | ALS/ALS + FTLD | Autophagosome recruitment to damaged mitochondria | 1%–4% | [35,87] |
| Ubiquilin-2 | Xp11.21 | X-linked dominant | ALS/ALS + FTLD | Substrate recognition and selective autophagy | <1% | [88] Reviewed in [89] |
| VCP/p97 (valosin-containing protein) | 9p13.3 | Dominant | ALS/ALS + FTLD | Autophagosome maturation | 1%–2% | [90] |
| Dynein/Dynactin | 2p13 | Dominant | ALS | Autophagosome transport and fusion with lysosome | Unknown | [91] |
| CHMP2B (Charged Multivesicular body protein 2b) | 3p11 | Dominant | ALS/ALS + FTLD | Autolysosome formation | Unknown | [47,92] |
| FIG4 (Phosphatidylinositol 3,5-bisphosphate 5-phosphatase) | 6q21 | Dominant | ALS | Alteration of endo-lysosomal trafficking | Unknown | [93] |
| ALS2 (Alsin) | 2q33.2 | Recessive | ALS | Alteration of endosomal trafficking | <1% | [94] |
| C9ORF72 | 9p21.3-p13.3 | Dominant | ALS + FTLD | Alteration of endosomal trafficking | 40% | [95,96,97] |
- -
- Dominant missense, recessive deletions and nonsense mutations of optineurin have been identified in both patients with FALS and SALS [87]. The relevance of optineurin mutations to ALS include impairment of the recruitment of damaged mitochondria to autophagosomes, as well as sequestration of functional proteins in hybrid complexes, which compromises the maturation of autophagosomes [35,56]. Furthermore, TDP-43- or SOD1-positive inclusions of sporadic and SOD1 cases of ALS have been found to contain also optineurin [87]. Interestingly, as in the case of p62, mutations of optineurin are also associated with PDB and, in addition, with glaucoma [33,34].
- -
- Mutations affecting the UBQLN2 gene were originally found as a cause of ALS by Deng et al. in 2011. Interestingly, ubiquitin pathology was not restricted to patients with ALS who had ubiquilin-2 mutations, but also to SALS and ALS cases with other mutations, in which UBQLN-2-positive inclusions could be identified in the brain and spinal cord [89]. Moreover, ubiquilin-2 (UBQLN2) binds with high affinity to TDP-43 and modulates TDP-43 levels. Given the presence of ubiquilin-2 in inclusions containing also other proteins (such as TDP43), UBQL2 has been suggested to play a role in ALS that may not be uniquely associated with its mutations [75]. Interestingly, a link between ALS-linked ubiquilin-2 and optineurin mutations has been recently proposed, since they both appear to interfere with the trafficking of endosomal vesicles, suggesting that these proteins may function in a common pathological process [38].
- -
- Mutations in the valosin-containing protein VCP/p97 lead to a multisystem degenerative disease consisting of inclusion body myopathy, early-onset Paget’s disease of the bone and fronto-temporal dementia (IBMPFD) [98]. Subsequently, mutations in VCP were found in a small percentage of familial ALS cases [90]. As discussed in the first section of this review, VCP is essential for ubiquitin-containing autophagosome maturation and endolysosomal sorting, as well as for establishing the fusion of lysosomes with autophagosomes; in addition, the expression of either disease-associated VCP mutants or dominant-negative VCP results in autophagy defects ([55] and the references therein). Very interestingly, VCP appears to play an important role in the autophagy-mediated degradation of stress granules, thus contributing to the formation of stress granule-based aggregates, a process that may indirectly impact RNA processing [42]. This function links VCP to two genes more commonly associated with FALS, which code for the RNA binding proteins TDP-43 and FUS (reviewed in [34,75]). An emerging idea is that the changes in RNA processing observed in ALS-FLTD may be the result of pathological stress granule formation, normally requiring intact autophagy, lysosomes and VCP [99]. Further, other ALS-FTLD-associated gene products, such as profilin 1 and ataxin 2, are stress granule-associated proteins [100]. Finally, VCP mutations have also been shown to affect the nutrient sensing function of mTOR, the main regulator of starvation-induced autophagy, although the precise mechanism remains unclear (reviewed in [11]).
- -
- Another ALS-causing gene with a clear function in autophagy is dynactin 1 (DCTN1) [91]. Mutations that affect the dynein motor machinery impair autophagosome-lysosome fusion, leading to decreased autophagic clearance of aggregated proteins and enhanced toxicity, while motor neuron-specific knockdown of the dynactin 1 homologue in the nematode was shown to disrupt autophagosome transport and induce motor neuron degeneration ([55,101] and the references therein).
- -
- A rare mutation in the endosomal ESCRTIII-complex subunit CHMP2B, which, as mentioned before, is part of the ESCRT-complex machinery whose activity is required to maintain autophagic flux, was found in human patients with an autosomal dominant form of FTLD linked to chromosome 3. Additional alterations have been reported in association with ALS phenotypes, thus strengthening the idea of a common molecular pathology between ALS and FTLD [47,92].
- -
- The convergence of the autophagic pathway with the endocytic pathway is underlined by the finding that the polyphosphoinositide phosphatase-encoding gene FIG4 (also known as SAC3) is mutated in some cases of FALS [93]. Given the importance of its enzyme product in the regulation of the intracellular levels of phosphatidylinositol 3,5-biphosphate and its metabolite phosphatidylinositol 3 phosphate (PI3P), and, consequently, in the lipid composition of late endosomal/lysosomal membranes, mutant FIG4 is thought to interfere with the trafficking of these organelles and, presumably, with a proper autophagic function, thus implicating phosphoinositide metabolism in the pathogenesis of ALS [74,75]. Mutations in this gene are also responsible for a variant of the Charcot-Marie-Tooth disorder (CMT4), characterized by axonal degeneration [50].
- -
- Mutations in the ALS2 gene, which encodes the protein alsin, account for several recessive MND, including ALS2. Alsin is a guanine nucleotide exchange factor (GEF) for the small GTPase protein Rab5 and, through its activation, is involved in endosome fusion and trafficking, as well as neurite outgrowth [94], reviewed in [74]. More recently, pathogenic missense mutations of alsin were shown to impair activation of Rab5, resulting in disturbances in the formation of amphisomes in cultures cell models [102]; reviewed in [55].
- -
- It is noteworthy that the protein C9ORF72, encoded by the most commonly-mutated gene in ALS, has been recently found to be structurally related to a GDP/GTP exchange factor (GEF) that activates Rab-GTPases [103] and to co-localize with Rab proteins, including Rab7, in neuronal cell lines, primary cortical neurons and human spinal cord motor neurons of ALS patients [97]. Since the small GTPase Rab7 is needed for the maturation of autophagosomes to autolysosomes (Figure 1), these studies on the still undeciphered function of C9ORF72 suggest that, unexpectedly, it may play a role in endocytic trafficking and autophagy (reviewed in [5,77,81]).
- -
- Finally, to complete this overview, it should be reported that the loss of function of some ALS-linked proteins that are neither bona fide autophagy receptors/adaptors nor directly involved in the process of autophagy, can be associated with defective protein degradation. For example, a mutation in the transmembrane domain of the sigma receptor-1 protein (SigR1), an ER chaperone protein located at the mitochondria-ER interface, has been found to cause a form of juvenile ALS and FTLD [104,105], and more recently, depletion of SigR1 has been shown to be associated with defects in endosomal trafficking and accumulation of autophagic substrates [106]. Although this protein was originally described as a sigma-opioid receptor, it was later found to be a distinct non-opioid receptor that binds a wide range of ligands and with a crucial role in Ca++ signaling, neuronal function and survival [105,107].
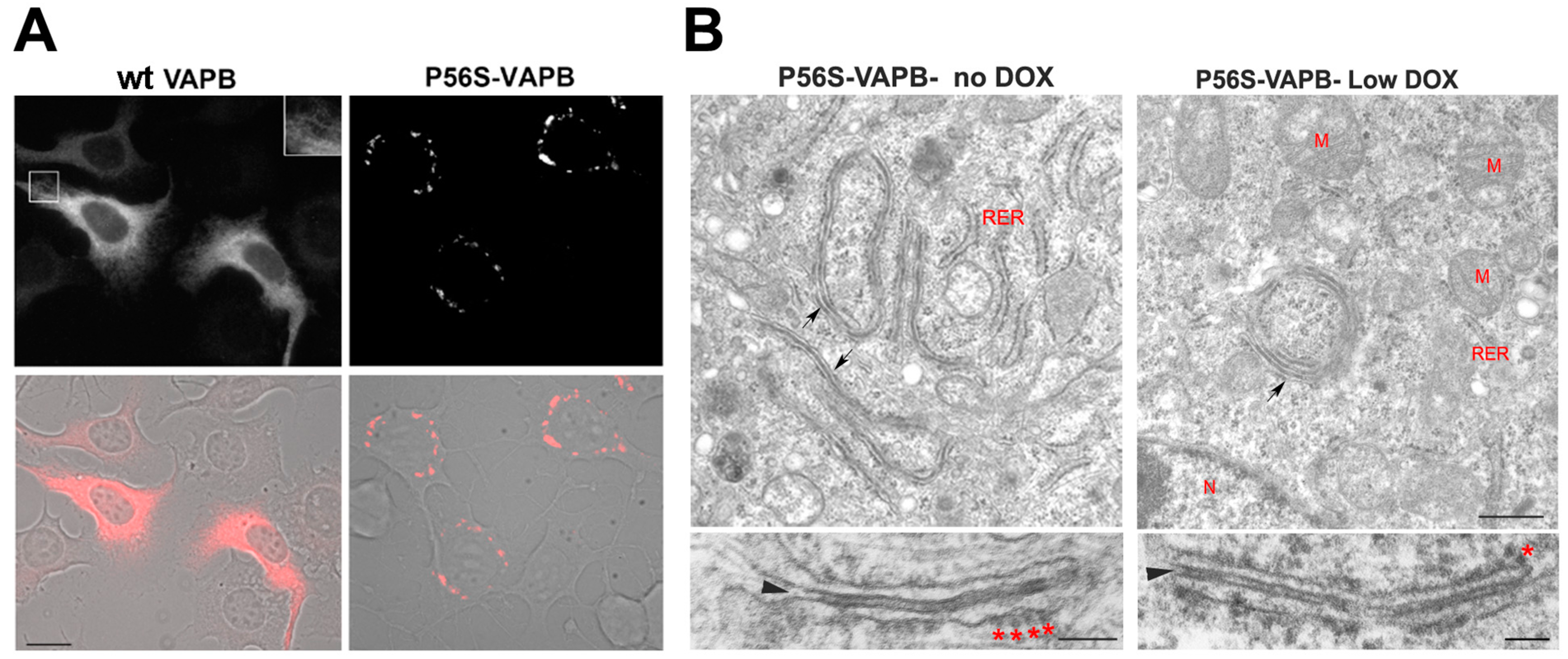
4. VAPB
5. Role of the VAPB Gene in the Pathogenesis of ALS

6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell. Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Invest. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim. Biophys. Acta. 2015, 1852, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian macroautophagy at a glance. J. Cell. Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Baskaran, S.; Schekman, R.; Hurley, J.H. The protein-vesicle network of autophagy. Curr. Opin. Cell. Biol. 2014, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Damme, M.; Suntio, T.; Saftig, P.; Eskelinen, E.L. Autophagy in neuronal cells: General principles and physiological and pathological functions. Acta. Neuropathol. 2015, 129, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell. Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell. Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell. 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell. Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell. Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Autophagy: Links with the proteasome. Curr. Opin. Cell. Biol. 2010, 22, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell. Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell. Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z. Neuronal aggregates: Formation, clearance, and spreading. Dev. Cell. 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell. Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell. Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T.; Wooten, M.W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 2007, 32, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Majcher, V.; Searle, M.S.; Layfield, R. SQSTM1 mutations--bridging Paget disease of bone and ALS/FTLD. Exp. Cell. Res. 2014, 325, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kachaner, D.; Genin, P.; Laplantine, E.; Weil, R. Toward an integrative view of Optineurin functions. Cell. Cycle 2012, 11, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Majcher, V.; Goode, A.; James, V.; Layfield, R. Autophagy receptor defects and ALS-FTLD. Mol. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, P.; Gao, H.; Gu, Y.; Yang, J.; Peng, H.; Xu, X.; Wang, H.; Yang, M.; Liu, X.; et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014, 26, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, C.; Monteiro, M.J. Ubiquilin at a crossroads in protein degradation pathways. Autophagy 2010, 6, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Osaka, M.; Ito, D.; Yagi, T.; Nihei, Y.; Suzuki, N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.J.; Danner, R.; Liang, J.; Doong, H.; Harman, C.; Srinivasan, D.; Rothenberg, C.; Wang, H.; Ye, Y.; Fang, S.; et al. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J. Cell. Biol. 2009, 187, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell. Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell. Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell. Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohsumi, Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007, 581, 2156–2161. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.; de Camilli, P. Phosphoinositides’ link to neurodegeneration. Nat. Med. 2007, 13, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Moreau, K.; Rubinsztein, D.C. Protein misfolding disorders and macroautophagy. Curr. Opin. Cell. Biol. 2011, 23, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell. Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Schiavo, G. Liaisons dangereuses: Autophagy, neuronal survival and neurodegeneration. Curr. Opin. Neurobiol. 2008, 18, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Autophagosome dynamics in neurodegeneration at a glance. J. Cell. Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Autophagosome assembly and cargo capture in the distal axon. Autophagy 2012, 8, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Sass, M.; Klionsky, D.J. The regulation of aging: Does autophagy underlie longevity? Trends Cell. Biol. 2009, 19, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Tan, M.S.; Jiang, T.; Zhu, X.C.; Tan, L. Autophagy in aging and neurodegenerative diseases: Implications for pathogenesis and therapy. Neurobiol. Aging 2014, 35, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell. Stem Cell. 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.T.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell. Stem Cell. 2014, 14, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell. Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Medinas, D.B.; Hetz, C. Common ground: Stem cell approaches find shared pathways underlying ALS. Cell. Stem Cell. 2014, 14, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy in neurodegenerative disease: Friend, foe or turncoat? Trends Neurosci. 2006, 29, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Mitochondria, autophagy and age-associated neurodegenerative diseases: New insights into a complex interplay. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Bennion Callister, J.; Pickering-Brown, S.M. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp. Neurol. 2014, 262, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Doucette, P.A.; Zittin Potter, S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef] [PubMed]
- Laferriere, F.; Polymenidou, M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med. Wkly. 2015, 145, w14054. [Google Scholar] [CrossRef] [PubMed]
- Boillee, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell. Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta. Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, X.; Song, L.; Le, W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012, 22, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Yang, S.; Warraich, S.T.; Blair, I.P. Ubiquilin 2: A component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell. Biol. 2014, 50, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Seguin, S.J.; Morelli, F.F.; Vinet, J.; Amore, D.; de Biasi, S.; Poletti, A.; Rubinsztein, D.C.; Carra, S. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell. Death Differ. 2014, 21, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Figley, M.D.; Bieri, G.; Kolaitis, R.M.; Taylor, J.P.; Gitler, A.D. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 2014, 34, 8083–8097. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Kawai, K.; Katsuno, M.; Huang, Z.; Jiang, Y.M.; Iguchi, Y.; Kobayashi, K.; Kimata, T.; Waza, M.; Tanaka, F.; et al. dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS ONE 2013, 8, e54511. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Ikeda, J.E.; Hadano, S. Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett. 2011, 585, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Prause, J.; Goswami, A.; Katona, I.; Roos, A.; Schnizler, M.; Bushuven, E.; Dreier, A.; Buchkremer, S.; Johann, S.; Beyer, C.; et al. Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 1581–1600. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell. Death Dis 2014, 5, e1290. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Shih, Y.T.; Chen, C.Y.; Chao, H.W.; Lee, M.J.; Hsueh, Y.P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Invest. 2011, 121, 4820–4837. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.P. From neurodevelopment to neurodegeneration: The interaction of neurofibromin and valosin-containing protein/p97 in regulation of dendritic spine formation. J. Biomed. Sci. 2012, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, Y.; Urushitani, M.; Inoue, H.; Koike, M.; Uchiyama, Y.; Komatsu, M.; Tanaka, K.; Yamazaki, M.; Abe, M.; Misawa, H.; et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 2012, 287, 42984–42994. [Google Scholar] [CrossRef] [PubMed]
- Lev, S.; Ben Halevy, D.; Peretti, D.; Dahan, N. The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol. 2008, 18, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Leal, S.N.S.; Ben Halevy, D.; Gomes, C.U.M.; Lev, S. Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 2010, 285, 13839–13849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huh, S.U.; Drennan, J.M.; Kathuria, H.; Martinez, J.S.; Tsuda, H.; Hall, M.C.; Clemens, J.C. Drosophila Vap-33 is required for axonal localization of Dscam isoforms. J. Neurosci. 2012, 32, 17241–17250. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.; van Dis, V.; Haasdijk, E.D.; Harterink, M.; Vocking, K.; Post, J.A.; Scheper, W.; Hoogenraad, C.C.; Jaarsma, D. Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta. Neuropathol. Commun. 2013, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Peretti, D.; Dahan, N.; Shimoni, E.; Hirschberg, K.; Lev, S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Boil. Cell 2008, 19, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Amarilio, R.; Ramachandran, S.; Sabanay, H.; Lev, S. Differential regulation of endoplasmic reticulum structure through VAP-Nir protein interaction. J. Biol. Chem. 2005, 280, 5934–5944. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.; Withers, J.; Koh, Y.H.; Parry, K.; Bao, H.; Zhang, B.; Budnik, V.; Pennetta, G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 2008, 17, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Shirane, M.; Natume, T.; Iemura, S.; Nakayama, K.I. Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J. Biol. Chem. 2009, 284, 13766–13777. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Shirane, M.; Hashimoto, Y.; Saita, S.; Nakayama, K.I. Identification and characterization of a neuron-specific isoform of protrudin. Genes Cells 2014, 19, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Suzuki, H.; Aiso, S.; Matsuoka, M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol. Neurobiol. 2009, 39, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.; Loewen, C. Inter-organelle membrane contact sites: Through a glass, darkly. Curr. Opin. Cell. Biol. 2006, 18, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell. Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Alpy, F.; Rousseau, A.; Schwab, Y.; Legueux, F.; Stoll, I.; Wendling, C.; Spiegelhalter, C.; Kessler, P.; Mathelin, C.; Rio, M.C.; et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell. Sci. 2013, 126, 5500–5512. [Google Scholar] [CrossRef] [PubMed]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; Macgurn, J.A.; Emr, S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell. 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Agaisse, H.; Derre, I. Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect. Immun. 2014, 82, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.; Middleton, S.; Kremer, A.M.; Wardrope, C.; Hannah, M.; Gillingwater, T.H.; Skehel, P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008, 17, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Kumagai, K.; Nishijima, M.; Hanada, K. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 2006, 281, 30279–30288. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J.; Manford, A.G.; Emr, S.D. ER-PM connections: Sites of information transfer and inter-organelle communication. Curr. Opin. Cell. Biol. 2013, 25, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, B.; Bigay, J.; Moser von Filseck, J.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013, 155, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.; Chai, A.; Sanhueza, M.; Marescotti, M.; Parry, K.; Georgiev, A.; Sahota, V.; Mendez-Castro, R.; Pennetta, G. Increased levels of phosphoinositides cause neurodegeneration in a Drosophila model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 2689–2704. [Google Scholar] [CrossRef] [PubMed]
- Mikitova, V.; Levine, T.P. Analysis of the key elements of FFAT-like motifs identifies new proteins that potentially bind VAP on the ER, including two AKAPs and FAPP2. PLoS ONE 2012, 7, e30455. [Google Scholar] [CrossRef] [PubMed]
- Baron, Y.; Pedrioli, P.G.; Tyagi, K.; Johnson, C.; Wood, N.T.; Fountaine, D.; Wightman, M.; Alexandru, G. VAPB/ALS8 interacts with FFAT-like proteins including the p97 cofactor FAF1 and the ASNA1 ATPase. BMC Biol. 2014, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Hantan, D.; Yamamoto, Y.; Sakisaka, T. VAP-B binds to Rab3GAP1 at the ER: Its implication in nuclear envelope formation through the ER-Golgi intermediate compartment. Kobe J. Med. Sci. 2014, 60, E48–E56. [Google Scholar] [PubMed]
- Spang, N.; Feldmann, A.; Huesmann, H.; Bekbulat, F.; Schmitt, V.; Hiebel, C.; Koziollek-Drechsler, I.; Clement, A.M.; Moosmann, B.; Jung, J.; et al. RAB3GAP1 and RAB3GAP2 modulate basal and rapamycin-induced autophagy. Autophagy 2014, 10, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Tsuda, H.; Yang, Y.; Vibbert, J.; Cottee, P.; Lee, S.-J.; Winek, J.; Haueter, C.; Bellen, H.J.; Miller, M.A. Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev. Cell 2012, 22, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.N.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.O.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Al-Chalabi, A.; Zatz, M. A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum. Genet. 2005, 118, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Funke, A.D.; Esser, M.; Krüttgen, A.; Weis, J.; Mitne-Neto, M.; Lazar, M.; Nishimura, A.L.; Sperfeld, A.D.; Trillenberg, P.; Senderek, J.; et al. The p.P56S mutation in the VAPB gene is not due to a single founder: The first European case. Clin. Genet. 2010, 77, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; van Es, M.A.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, 2950-e1. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, M.; Zechini, L.; Gillespie, T.; Pennetta, G. Gain-of-function mutations in the ALS8 causative gene VAPB have detrimental effects on neurons and muscles. Biol. Open 2014, 3, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; El Oussini, H.; Bercier, V.; Gros-Louis, F.; Valdmanis, P.N.; McDearmid, J.; Mejier, I.A.; Dion, P.A.; Dupre, N.; Hollinger, D.; et al. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.O.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci.: Off. J. Soc. Neurosci. 2007, 27, 9801–9815. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Lua, S.; Tong, J.S.; Song, J. Elimination of the native structure and solubility of the hVAPB MSP domain by the Pro56Ser mutation that causes amyotrophic lateral sclerosis. Biochemistry 2010, 49, 3887–3897. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, G.; Akbar, M.T.; Paul, P.; Angelinetta, C.; Steiner, T.J.; de Belleroche, J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 2010, 31, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Langou, K.; Moumen, A.; Pellegrino, C.; Aebischer, J.; Medina, I.; Aebischer, P.; Raoul, C. AAV-mediated expression of wild-type and ALS-linked mutant VAPB selectively triggers death of motoneurons through a Ca2+-dependent ER-associated pathway. J. Neurochem. 2010, 114, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Matsuoka, M. Amyotrophic lateral sclerosis-linked mutant VAPB enhances TDP-43-induced motor neuronal toxicity. J. Neurochem. 2011, 119, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Mόrotz, G.M.; de Vos, K.J.; Vagnoni, A.; Ackerley, S.; Shaw, C.E.; Miller, C.C.J. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 2012, 21, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Ratnaparkhi, A.; Lawless, G.M.; Schweizer, F.E.; Golshani, P.; Jackson, G.R. A Drosophila model of ALS: Human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE 2008, 3, e2334. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kanekura, K.; Levine, T.P.; Kohno, K.; Olkkonen, V.M.; Aiso, S.; Matsuoka, M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009, 108, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, L.; Lai, C.; Yu, J.; Chub, N.; Shim, H.; Sun, L.; Xie, C.; Yang, W.J.; Lin, X.; O’Donovan, M.J.; et al. Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum. Mol. Genet. 2013, 22, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Moustaqim-Barrette, A.; Lin, Y.Q.; Pradhan, S.; Neely, G.G.; Bellen, H.J.; Tsuda, H. The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum. Mol. Genet. 2014, 23, 1975–1989. [Google Scholar] [CrossRef] [PubMed]
- Deivasigamani, S.; Verma, H.K.; Ueda, R.; Ratnaparkhi, A.; Ratnaparkhi, G.S. A genetic screen identifies Tor as an interactor of VAPB in a Drosophila model of amyotrophic lateral sclerosis. Biol. Open 2014, 3, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Papiani, G.; Ruggiano, A.; Fossati, M.; Raimondi, A.; Bertoni, G.; Francolini, M.; Benfante, R.; Navone, F.; Borgese, N. Restructured Endoplasmic Reticulum, Generated by Mutant, Amyotrophic Lateral Sclerosis-Linked VAPB, is Cleared by the Proteasome. J. Cell Sci. 2012, 125, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Genevini, P.; Papiani, G.; Ruggiano, A.; Cantoni, L.; Navone, F.; Borgese, N. Amyotrophic lateral sclerosis-linked mutant VAPB inclusions do not interfere with protein degradation pathways or intracellular transport in a cultured cell model. PLoS ONE 2014, 9, e113416. [Google Scholar] [CrossRef] [PubMed]
- Snapp, E.L.; Hegde, R.S.; Francolini, M.; Lombardo, F.; Colombo, S.; Pedrazzini, E.; Borgese, N.; Lippincott-Schwartz, J. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell. Biol. 2003, 163, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Mignery, G.A.; Mugnaini, E.; Sudhof, T.C.; de Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron 1994, 12, 327–342. [Google Scholar] [CrossRef]
- Mitne-Neto, M.; Machado-Costa, M.; Marchetto, M.C.; Bengtson, M.H.; Joazeiro, C.A.; Tsuda, H.; Bellen, H.J.; Silva, H.C.; Oliveira, A.S.; Lazar, M.; et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 2011, 20, 3642–3652. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Virard, I.; Raoul, C. Accumulation of wildtype and ALS-linked mutated VAPB impairs activity of the proteasome. PLoS ONE 2011, 6, e26066. [Google Scholar] [CrossRef] [PubMed]
- Prosser, D.C.; Tran, D.; Gougeon, P.Y.; Verly, C.; Ngsee, J.K. FFAT rescues VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J. Cell. Sci. 2008, 121, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Song, W.; Jiang, A.; Shyr, Y.; Lev, S.; Greenstein, D.; Brantley-Sieders, D.; Chen, J. VAMP-associated protein B (VAPB) promotes breast tumor growth by modulation of Akt activity. PLoS ONE 2012, 7, e46281. [Google Scholar] [CrossRef] [PubMed]
- Tudor, E.L.; Galtrey, C.M.; Perkinton, M.S.; Lau, K.F.; de Vos, K.J.; Mitchell, J.C.; Ackerley, S.; Hortobagyi, T.; Vamos, E.; Leigh, P.N.; et al. Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience 2010, 167, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Qiao, T.; Beers, M.; Tan, W.; Wang, H.; Yang, B.; Xu, Z. Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice. Mol. Neurodegener. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; El Oussini, H.; Scekic-Zahirovic, J.; Vibbert, J.; Cottee, P.; Prasain, J.K.; Bellen, H.J.; Dupuis, L.; Miller, M.A. VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 2013, 9, e1003738. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navone, F.; Genevini, P.; Borgese, N. Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS. Cells 2015, 4, 354-386. https://doi.org/10.3390/cells4030354
Navone F, Genevini P, Borgese N. Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS. Cells. 2015; 4(3):354-386. https://doi.org/10.3390/cells4030354
Chicago/Turabian StyleNavone, Francesca, Paola Genevini, and Nica Borgese. 2015. "Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS" Cells 4, no. 3: 354-386. https://doi.org/10.3390/cells4030354
APA StyleNavone, F., Genevini, P., & Borgese, N. (2015). Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS. Cells, 4(3), 354-386. https://doi.org/10.3390/cells4030354