Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression

Abstract
:1. Introduction
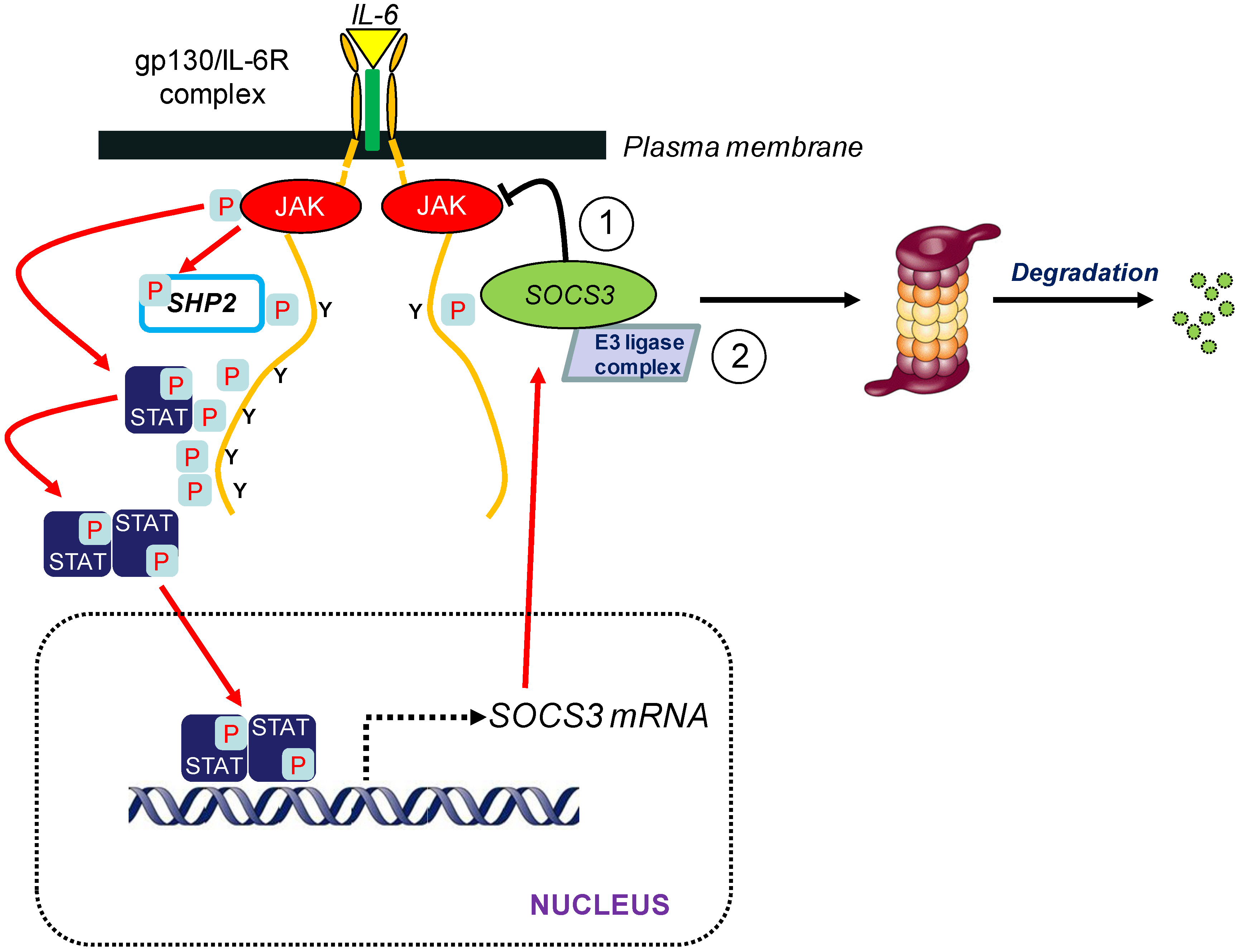
2. Suppressor of Cytokine Signalling 3 (SOCS3)
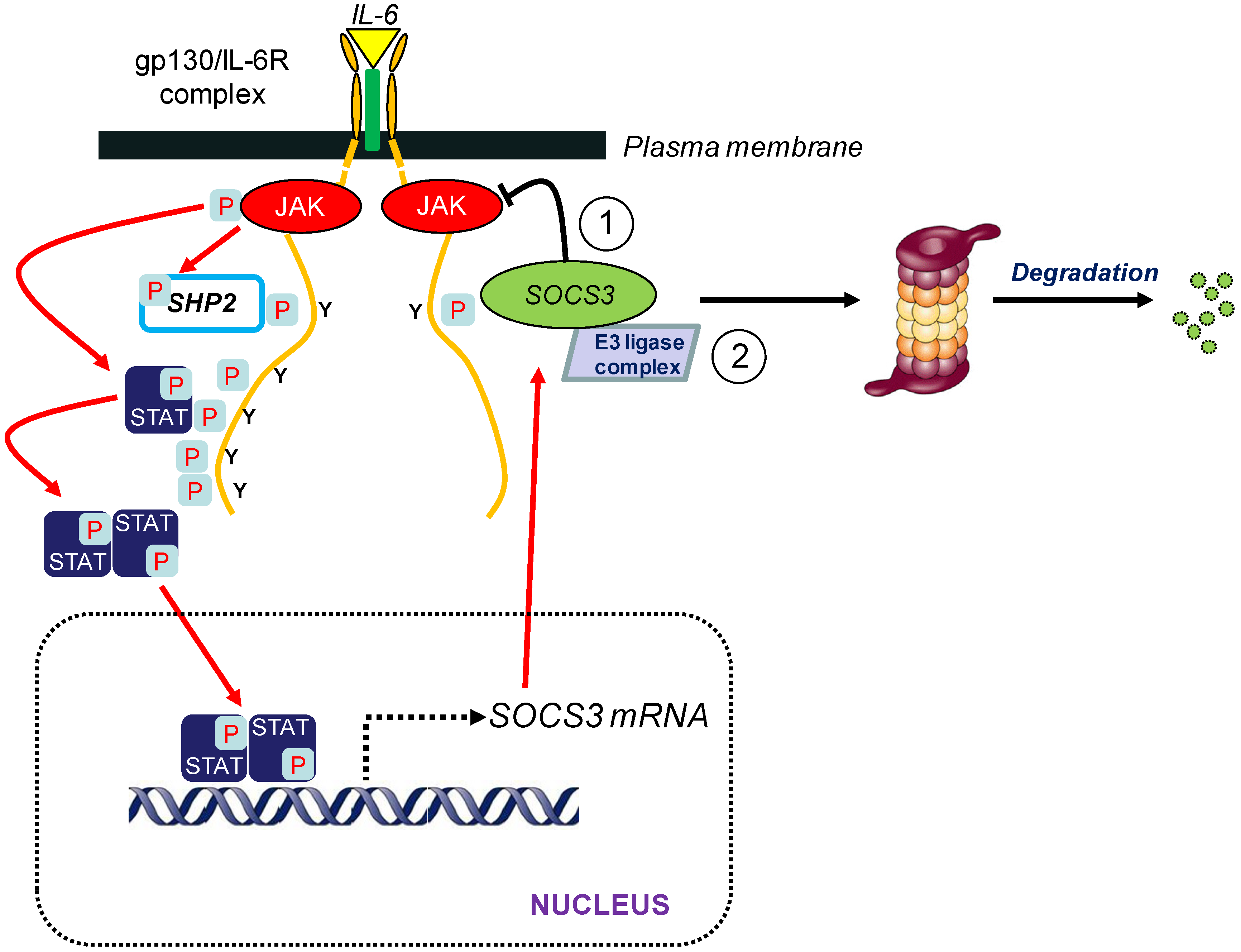
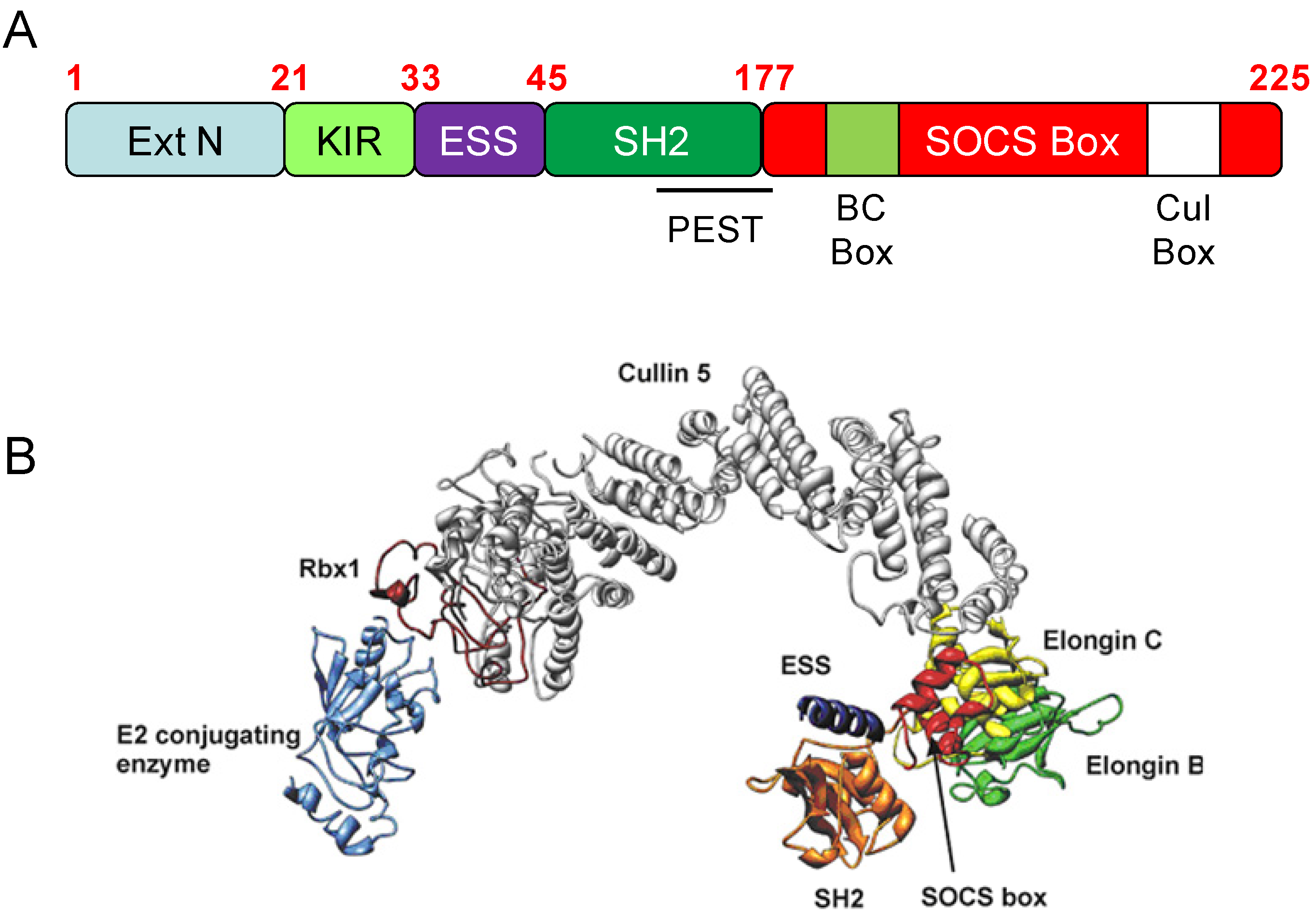
3. SOCS3 and E3 Ubiquitin Ligase Activity
3.1. Introduction
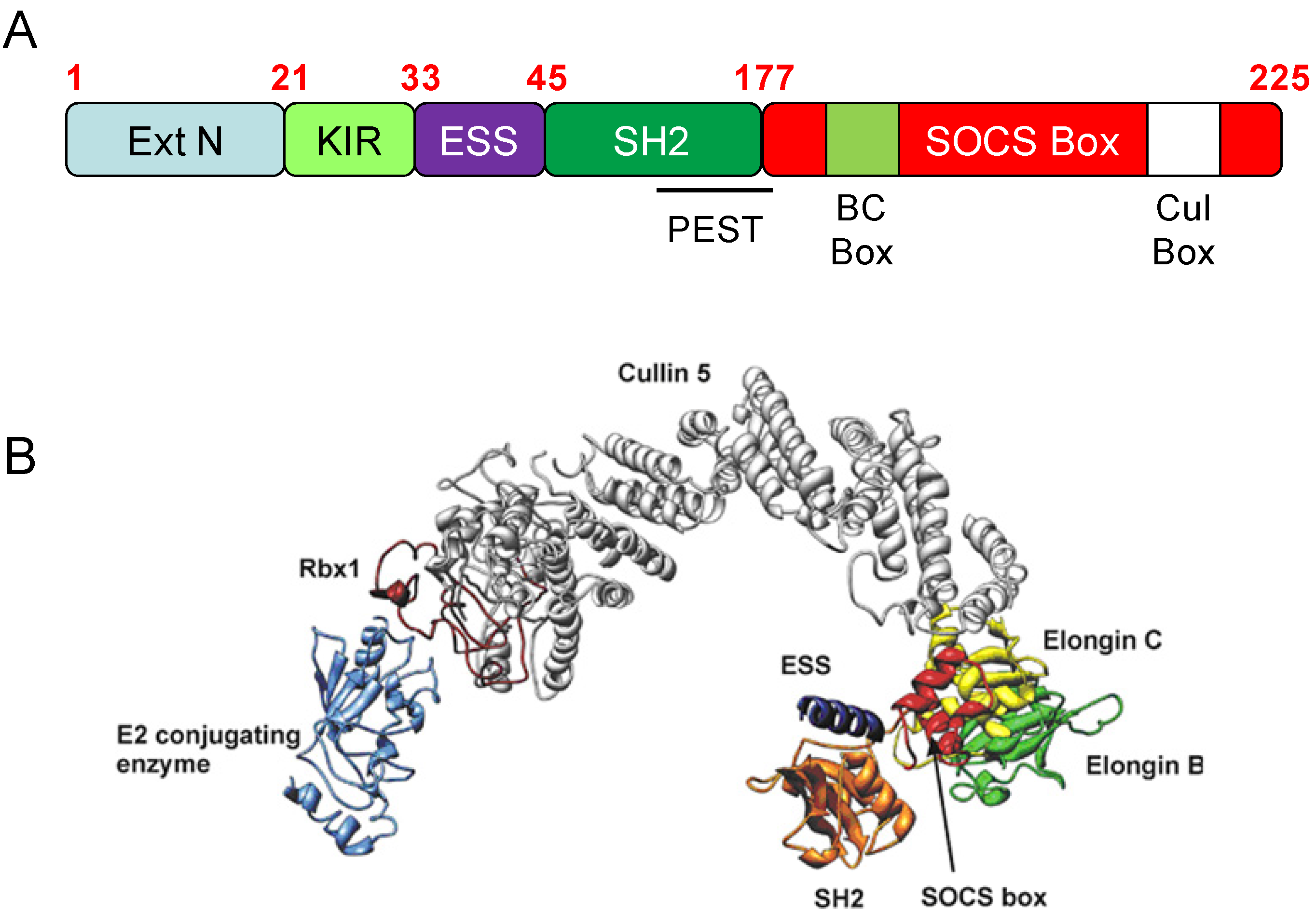

{kind=link}
{kind=link}
| Substrate | Ref. | |
|---|---|---|
| 1 | Janus kinase 1 (JAK1) | [29] |
| 2 | Focal adhesion kinase 1 (FAK1) | [30] |
| 3 | Insulin receptor substrate 1/2 (IRS1/2) | [31,32] |
| 4 | Sialic acid-binding Ig-like lectin (SIGLEC) 3/CD33 | [33] |
| 5 | Sialic acid-binding Ig-like lectin (SIGLEC) 7 | [34] |
| 6 | Indoleamine 2,3-dioxygenase (IDO) | [35] |
| 7 | Granulocyte colony-stimulating factor receptor (G-CSFR) | [36] |
3.2. Putative Substrates for SOCS3-Mediated Ubiquitylation
3.2.1. Indoleamine 2,3-Dioxygenase (IDO)
3.2.2. Focal Adhesion Kinase 1 (FAK1)
3.2.3. Insulin Receptor Substrate (IRS) 1 and 2
3.2.4. Sialic acid-binding Ig-like lectin (SIGLEC) 3 and 7
3.2.5. Granulocyte Colony-Stimulating Factor Receptor (G-CSFR)
4. Regulation of SOCS3 Turnover
4.1. Introduction
4.2. SOCS3 Tyr Phosphorylation and Degradation
4.3. SOCS2 as a Regulator of SOCS3 Stability and Turnover
5. Future Directions
Acknowledgments
Author Contributions
Conflict of Interest
References
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Eulenfeld, R.; Dittrich, A.; Khouri, C.; Müller, P.J.; Mütze, B.; Wolf, A.; Schaper, F. Interleukin-6 signalling: More than JAKs and STATs. Eur. J. Cell. Biol. 2012, 91, 486–495. [Google Scholar] [CrossRef]
- Fonseca, J.E.; Santos, M.J.; Canhão, H.; Choy, E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun. Rev. 2009, 8, 538–542. [Google Scholar] [CrossRef]
- Ortiz-Muñoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Muñoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef]
- Tibes, R.; Bogenberger, J.M.; Geyer, H.L.; Mesa, R.A. JAK2 inhibitors in the treatment of myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2012, 21, 1755–1774. [Google Scholar] [CrossRef]
- Ishimoto, H. Epigenetic alterations in cholangiocarcinoma-sustained IL-6/STAT3 signaling in cholangio-carcinoma due to SOCS3 epigenetic silencing. Digestion 2009, 79, 2–8. [Google Scholar] [CrossRef]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 2011, 121, 3375–3383. [Google Scholar] [CrossRef]
- Linossi, E.M.; Babon, J.J.; Hilton, D.J.; Nicholson, S.E. Suppression of cytokine signaling: The SOCS perspective. Cytokine Growth F. R. 2013, 24, 241–248. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Naka, T.; Fujimoto, M.; Tsutsui, H.; Yoshimura, A. Negative regulation of cytokine and TLR signalings by SOCS and others. Adv. Immunol. 2005, 87, 61–122. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Greenhalgh, C.J.; Norstedt, G.; Rico-Bautista, E. Negative regulation of growth hormone receptor signaling. Mol. Endocrinol. 2006, 20, 241–253. [Google Scholar] [CrossRef]
- Sands, W.A.; Woolson, H.D.; Milne, G.R.; Rutherford, C.; Palmer, T.M. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol. Cell Biol. 2006, 26, 6333–6346. [Google Scholar] [CrossRef]
- Nicholson, S.E.; de Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6493–6498. [Google Scholar] [CrossRef]
- De Souza, D.; Fabri, L.J.; Nash, A.; Hilton, D.J.; Nicola, N.A.; Baca, M. SH2 domains from suppressor of cytokine signaling-3 and protein tyrosine phosphatase SHP-2 have similar binding specificities. Biochemistry 2002, 41, 9229–9236. [Google Scholar] [CrossRef]
- Lehmann, U.; Schmitz, J.; Weissenbach, M.; Sobota, R.M.; Hortner, M.; Friederichs, K.; Behrmann, I.; Tsiaris, W.; Sasaki, A.; Schneider-Mergener, J.; et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J. Biol. Chem. 2003, 278, 661–671. [Google Scholar]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef]
- Nicholson, S.E.; Willson, T.A.; Farley, A.; Starr, R.; Zhang, J.G.; Baca, M.; Alexander, W.S.; Metcalf, D.; Hilton, D.J.; Nicola, N.A. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999, 18, 375–385. [Google Scholar] [CrossRef]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef]
- Lucet, I.S.; Fantino, E.; Styles, M.; Bamert, R.; Patel, O.; Broughton, S.E.; Walter, M.; Burns, C.J.; Treutlein, H.; Wilks, A.F.; et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood 2006, 107, 176–183. [Google Scholar] [CrossRef]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef]
- Johnston, J.A. Are SOCS suppressors, regulators, and degraders? J. Leukocyte Biol. 2004, 75, 743–748. [Google Scholar] [CrossRef]
- White, C.A.; Nicola, N.A. SOCS3: An essential physiological inhibitor of signaling by interleukin-6 and G-CSF family cytokines. Jakstat 2013, 2, e25045. [Google Scholar] [CrossRef]
- Babon, J.J.; Sabo, J.K.; Zhang, J.G.; Nicola, N.A.; Norton, R.S. The SOCS box encodes a hierarchy of affinities for Cullin5: Implications for ubiquitin ligase formation and cytokine signalling suppression. J. Mol. Biol. 2009, 387, 162–174. [Google Scholar] [CrossRef]
- Zhang, J.G.; Metcalf, D.; Rakar, S.; Asimakis, M.; Greenhalgh, C.J.; Willson, T.A.; Starr, R.; Nicholson, S.E.; Carter, W.; Alexander, W.S.; et al. The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 13261–13265. [Google Scholar] [CrossRef]
- Boyle, K.; Egan, P.; Rakar, S.; Willson, T.A.; Wicks, I.P.; Metcalf, D.; Hilton, D.J.; Nicola, N.A.; Alexander, W.S.; Roberts, A.W.; et al. The SOCS box of suppressor of cytokine signaling-3 contributes to the control of G-CSF responsiveness in vivo. Blood 2007, 110, 1466–1474. [Google Scholar] [CrossRef]
- Piessevaux, J.; Lavens, D.; Peelman, F.; Tavernier, J. The many faces of the SOCS box. Cytokine Growth F. R. 2008, 19, 371–381. [Google Scholar] [CrossRef]
- Boyle, K.; Zhang, J.G.; Nicholson, S.E.; Trounson, E.; Babon, J.J.; McManus, E.J.; Nicola, N.A.; Robb, L. Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal. 2009, 21, 394–404. [Google Scholar] [CrossRef]
- Liu, E.; Cote, J.F.; Vuori, K. Negative regulation of FAK signaling by SOCS proteins. EMBO J. 2003, 22, 5036–5046. [Google Scholar] [CrossRef]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar]
- Kawaguchi, T.; Yoshida, T.; Harada, M.; Hisamoto, T.; Nagao, Y.; Ide, T.; Taniguchi, E.; Kumemura, H.; Hanada, S.; Maeyama, M.; et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 2004, 165, 1499–1508. [Google Scholar] [CrossRef]
- Orr, S.J.; Morgan, N.M.; Elliott, J.; Burrows, J.F.; Scott, C.J.; McVicar, D.W.; Johnston, J.A. CD33 responses are blocked by SOCS3 through accelerated proteasomal-mediated turnover. Blood 2007, 109, 1061–1068. [Google Scholar]
- Orr, S.J.; Morgan, N.M.; Buick, R.J.; Boyd, C.R.; Elliott, J.; Burrows, J.F.; Jefferies, C.A.; Crocker, P.R.; Johnston, J.A. SOCS3 targets Siglec 7 for proteasomal degradation and blocks Siglec 7-mediated responses. J. Biol. Chem. 2007, 282, 3418–3422. [Google Scholar]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Grohmann, U.; Puccetti, P.; Fallarino, F. Proteasomal degradation of indoleamine 2,3-dioxygenase in CD8 dendritic cells is mediated by suppressor of cytokine signaling 3 (SOCS3). Int. J. Tryptophan Res. 2010, 3, 91–97. [Google Scholar]
- Irandoust, M.I.; Aarts, L.H.; Roovers, O.; Gits, J.; Erkeland, S.J.; Touw, I.P. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 2007, 26, 1782–1793. [Google Scholar] [CrossRef]
- Ungureanu, D.; Saharinen, P.; Junttila, I.; Hilton, D.J.; Silvennoinen, O. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol. Cell. Biol. 2002, 22, 3316–3326. [Google Scholar] [CrossRef]
- Shortman, K.; Heath, W.R. The CD8+ dendritic cell subset. Immunol. Rev. 2010, 234, 18–31. [Google Scholar] [CrossRef]
- Lindemann, C.; Hackmann, O.; Delic, S.; Schmidt, N.; Reifenberger, G.; Riemenschneider, M.J. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 2011, 122, 241–251. [Google Scholar] [CrossRef]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar]
- Zhang, J.; Hochwald, S.N. The role of FAK in tumor metabolism and therapy. Pharmacol. Ther. 2013, 142, 154–163. [Google Scholar] [CrossRef]
- Seki, Y.; Inoue, H.; Nagata, N.; Hayashi, K.; Fukuyama, S.; Matsumoto, K.; Komine, O.; Hamano, S.; Himeno, K.; Inagaki-Ohara, K.; et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat. Med. 2003, 9, 1047–1054. [Google Scholar] [CrossRef]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Yamaji, T.; Mitsuki, M.; Teranishi, T.; Hashimoto, Y. Characterization of inhibitory signaling motifs of the natural killer cell receptor Siglec-7: Attenuated recruitment of phosphatases by the receptor is attributed to two amino acids in the motifs. Glycobiolog 2005, 15, 667–676. [Google Scholar] [CrossRef]
- Touw, I.P.; van de Geijn, G.J. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci. 2007, 12, 800–815. [Google Scholar] [CrossRef]
- Hörtner, M.; Nielsch, U.; Mayr, L.M.; Johnston, J.A.; Heinrich, P.C.; Haan, S. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J. Immunol. 2002, 169, 1219–1227. [Google Scholar] [CrossRef]
- Wölfler, A.; Irandoust, M.; Meenhuis, A.; Gits, J.; Roovers, O.; Touw, I.P. Site-specific ubiquitination determines lysosomal sorting and signal attenuation of the granulocyte colony-stimulating factor receptor. Traffic 2009, 10, 1168–1179. [Google Scholar] [CrossRef]
- Blanchard, F.; Wang, Y.; Kinzie, E.; Duplomb, L.; Godard, A.; Baumann, H. Oncostatin M regulates the synthesis and turnover of gp130, leukemia inhibitory factor receptor alpha, and oncostatin M receptor beta by distinct mechanisms. J. Biol. Chem. 2001, 276, 47038–47045. [Google Scholar]
- Thiel, S.; Dahmen, H.; Martens, A.; Müller-Newen, G.; Schaper, F.; Heinrich, P.C.; Graeve, L. Constitutive internalization and association with adaptor protein-2 of the interleukin-6 signal transducer gp130. FEBS Lett. 1998, 441, 231–234. [Google Scholar] [CrossRef]
- Fletcher, T.C.; DiGiandomenico, A.; Hawiger, J. Extended anti-inflammatory action of a degradation-resistant mutant of cell-penetrating suppressor of cytokine signaling 3. J. Biol. Chem. 2010, 285, 18727–18736. [Google Scholar] [CrossRef]
- Sasaki, A.; Inagaki-Ohara, K.; Yoshida, T.; Yamanaka, A.; Sasaki, M.; Yasukawa, H.; Koromilas, A.E.; Yoshimura, A. The N-terminal truncated isoform of SOCS3 translated from an alternative initiation AUG codon under stress conditions is stable due to the lack of a major ubiquitination site, Lys-6. J. Biol. Chem. 2003, 278, 2432–2436. [Google Scholar] [CrossRef]
- Babon, J.J.; McManus, E.J.; Yao, S.; DeSouza, D.P.; Mielke, L.A.; Sprigg, N.S.; Willson, T.A.; Hilton, D.J.; Nicola, N.A.; Baca, M.; et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol. Cell. 2006, 22, 205–216. [Google Scholar] [CrossRef]
- Haan, S.; Ferguson, P.; Sommer, U.; Hiremath, M.; McVicar, D.W.; Heinrich, P.C.; Johnston, J.A.; Cacalano, N.A. Tyrosine phosphorylation disrupts elongin interaction and accelerates SOCS3 degradation. J. Biol. Chem. 2003, 278, 31972–31979. [Google Scholar] [CrossRef]
- Sommer, U.; Schmid, C.; Sobota, R.M.; Lehmann, U.; Stevenson, N.J.; Johnston, J.A.; Schaper, F.; Heinrich, P.C.; Haan, S. Mechanisms of SOCS3 phosphorylation upon interleukin-6 stimulation. Contributions of Src- and receptor-tyrosine kinases. J. Biol. Chem. 2005, 280, 31478–31488. [Google Scholar] [CrossRef]
- Hookham, M.B.; Elliott, J.; Suessmuth, Y.; Staerk, J.; Ward, A.C.; Vainchenker, W.; Percy, M.J.; McMullin, M.F.; Constantinescu, S.N.; Johnston, J.A. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood 2007, 109, 4924–4929. [Google Scholar] [CrossRef]
- Haan, S.; Wüller, S.; Kaczor, J.; Rolvering, C.; Nöcker, T.; Behrmann, I.; Haan, C. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene 2009, 28, 3069–3080. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Elliott, J.; Barry, A.C.; Hibbert, L.; Cacalano, N.A.; Johnston, J.A. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol. Cell. Biol. 2005, 25, 9115–9126. [Google Scholar] [CrossRef]
- Piessevaux, J.; Lavens, D.; Montoye, T.; Wauman, J.; Catteeuw, D.; Vandekerckhove, J.; Belsham, D.; Peelman, F.; Tavernier, J. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J. Biol. Chem. 2006, 281, 32953–32966. [Google Scholar] [CrossRef]
- Spence, S.; Fitzsimons, A.; Boyd, C.R.; Kessler, J.; Fitzgerald, D.; Elliott, J.; Gabhann, J.N.; Smith, S.; Sica, A.; Hams, E.; et al. Suppressors of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity 2013, 38, 66–78. [Google Scholar] [CrossRef]
- Kiu, H.; Greenhalgh, C.J.; Thaus, A.; Hilton, D.J.; Nicola, N.A.; Alexander, W.S.; Roberts, A.W. Regulation of multiple cytokine signalling pathways by SOCS3 is independent of SOCS2. Growth Factors 2009, 27, 384–393. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Laurence, A.; McInnes, I.B. Back to the future: Oral targeted therapy for RA and other autoimmune diseases. Nat. Rev. Rheumatol. 2013, 9, 173–182. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Therapeutic targeting of the interleukin-6 receptor. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 199–219. [Google Scholar] [CrossRef]
- Serruys, P.W.; Kutryk, M.J.; Ong, A.T. Coronary-artery stents. N. Engl. J. Med. 2006, 354, 483–495. [Google Scholar] [CrossRef]
- Gupta, G.K.; Dhar, K.; del Core, M.G.; Hunter, W.J., III; Hatzoudis, G.I.; Agrawal, D.K. Suppressor of cytokine signaling-3 and intimal hyperplasia in porcine coronary arteries following coronary intervention. Exp. Mol. Pathol. 2011, 91, 346–352. [Google Scholar] [CrossRef]
- Dhar, K.; Rakesh, K.; Pankajakshan, D.; Agrawal, D.K. SOCS3 promotor hypermethylation and STAT3-NF-κB interaction downregulate SOCS3 expression in human coronary artery smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H776–H785. [Google Scholar] [CrossRef]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef]
- Nagata, T.; Kai, H.; Shibata, R.; Koga, M.; Yoshimura, A.; Imaizumi, T. Oncostatin M, an interleukin-6 family cytokine, upregulates matrix metalloproteinase-9 through the mitogen-activated protein kinase kinase-extracellular signal-regulated kinase pathway in cultured smooth muscle cells. Arterioscler Thromb. Vasc. Biol. 2003, 23, 588–593. [Google Scholar] [CrossRef]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Li, G.; Jin, R.; Norris, R.A.; Zhang, L.; Yu, S.; Wu, F.; Markwald, R.R.; Nanda, A.; Conway, S.J.; Smyth, S.S.; et al. Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 2010, 208, 358–365. [Google Scholar] [CrossRef]
- Shouda, T.; Yoshida, T.; Hanada, T.; Wakioka, T.; Oishi, M.; Miyoshi, K.; Komiya, S.; Kosai, K.; Hanakawa, Y.; Hashimoto, K.; et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Invest. 2001, 108, 1781–1788. [Google Scholar] [CrossRef]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar] [CrossRef]
- Xiang, S.; Liu, J.; Dong, N.; Shi, J.; Xiao, Y.; Wang, Y.; Hu, X.; Gong, L.; Wang, W. Suppressor of cytokine signaling 3 is a negative regulator for neointimal hyperplasia of vein graft stenosis. J. Vasc. Res. 2014, 51, 132–143. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Williams, J.J.L.; Munro, K.M.A.; Palmer, T.M. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells 2014, 3, 546-562. https://doi.org/10.3390/cells3020546
Williams JJL, Munro KMA, Palmer TM. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells. 2014; 3(2):546-562. https://doi.org/10.3390/cells3020546
Chicago/Turabian StyleWilliams, Jamie J. L., Kirsten M. A. Munro, and Timothy M. Palmer. 2014. "Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression" Cells 3, no. 2: 546-562. https://doi.org/10.3390/cells3020546
APA StyleWilliams, J. J. L., Munro, K. M. A., & Palmer, T. M. (2014). Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells, 3(2), 546-562. https://doi.org/10.3390/cells3020546