Imaging and Quantitation Techniques for Tracking Cargo along Endosome-to-Golgi Transport Pathways

{kind=link}
{kind=link}
Abstract
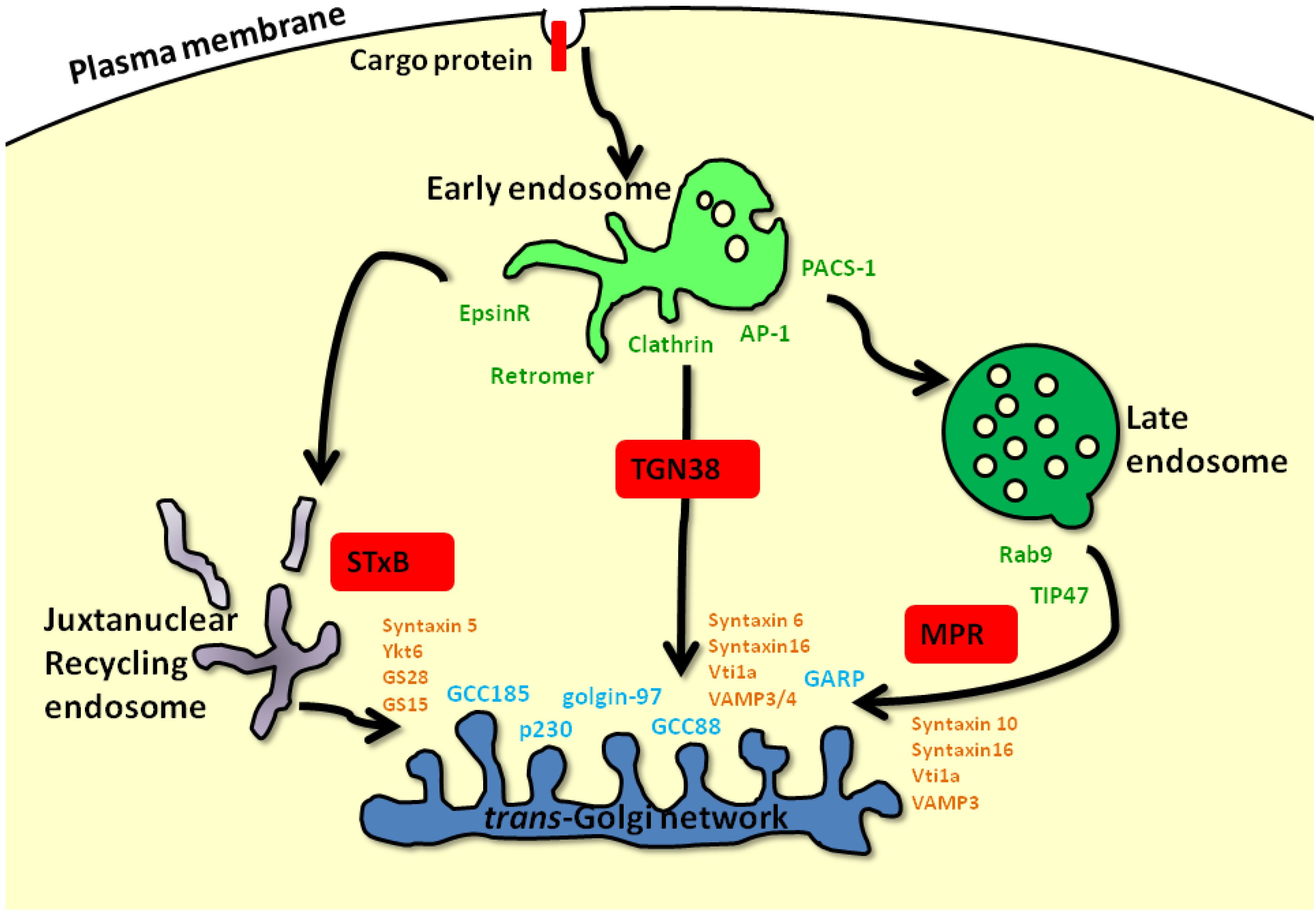
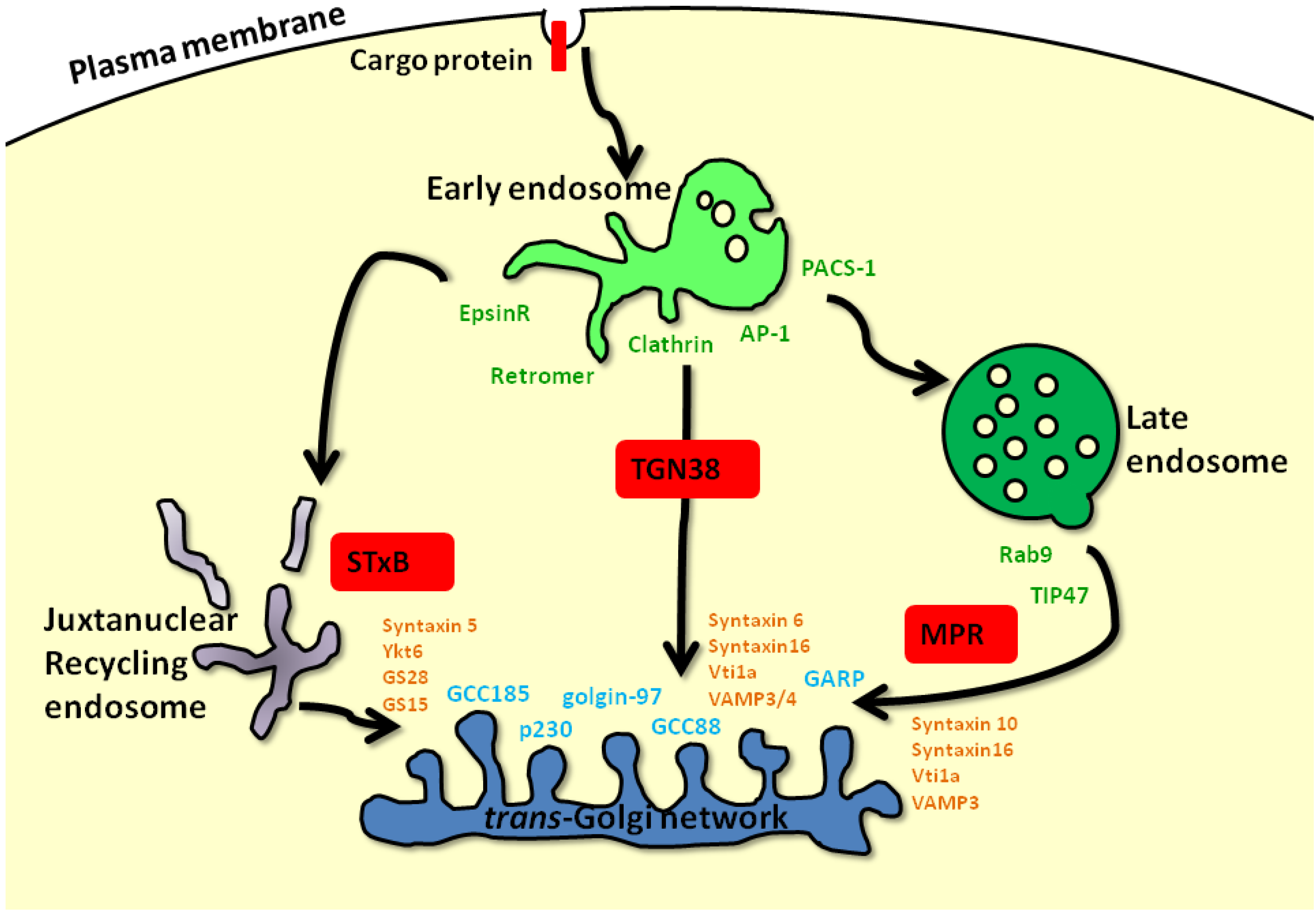
:1. Introduction: Intracellular Membrane Trafficking
2. Super-Resolution Light Microscopy: Resolving Nanomolecular Events
3. Correlative Light and Electron Microscopy
4. Recent Innovations in Fluorescent Proteins
5. Tools for the Analysis of Colocalization
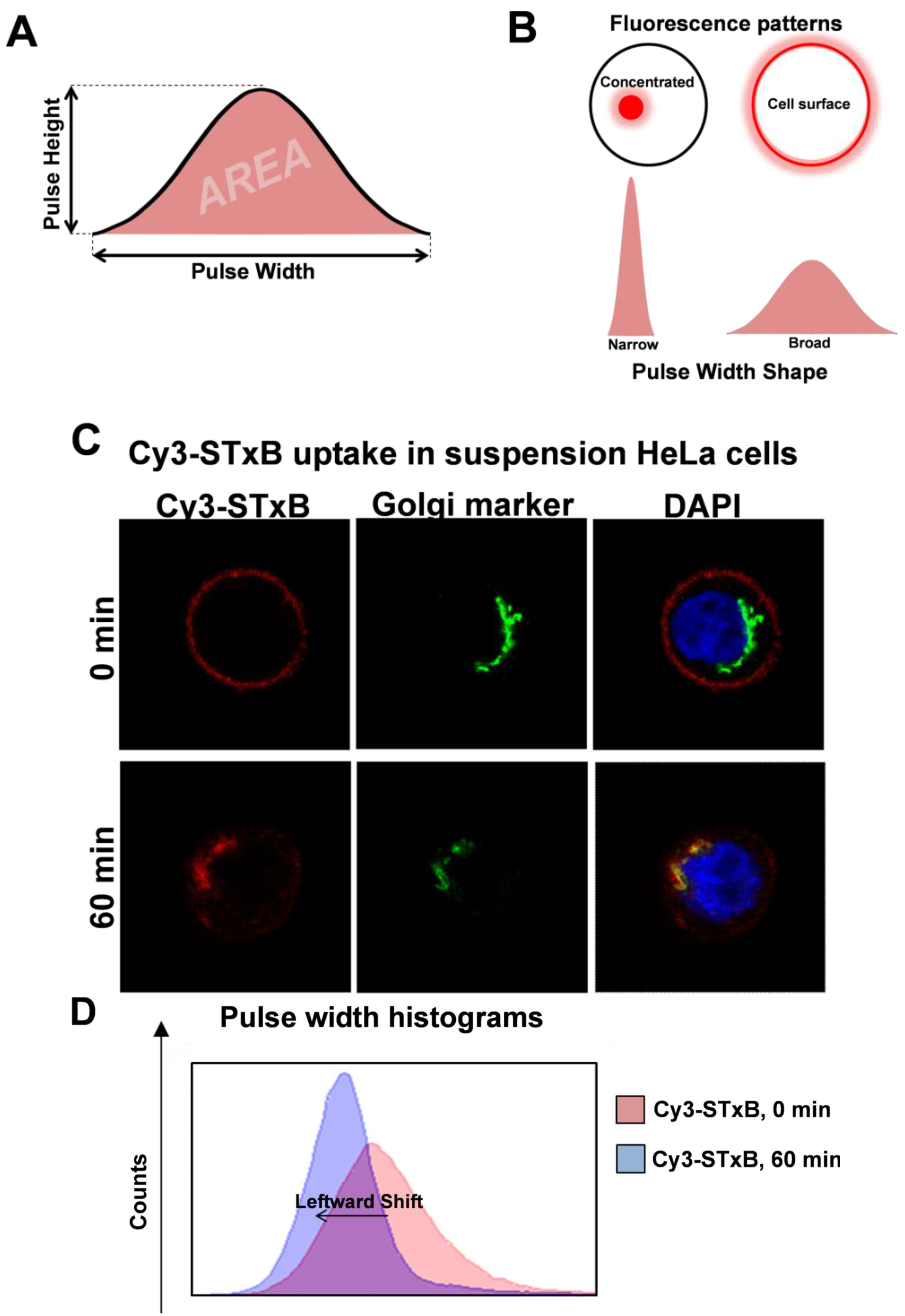
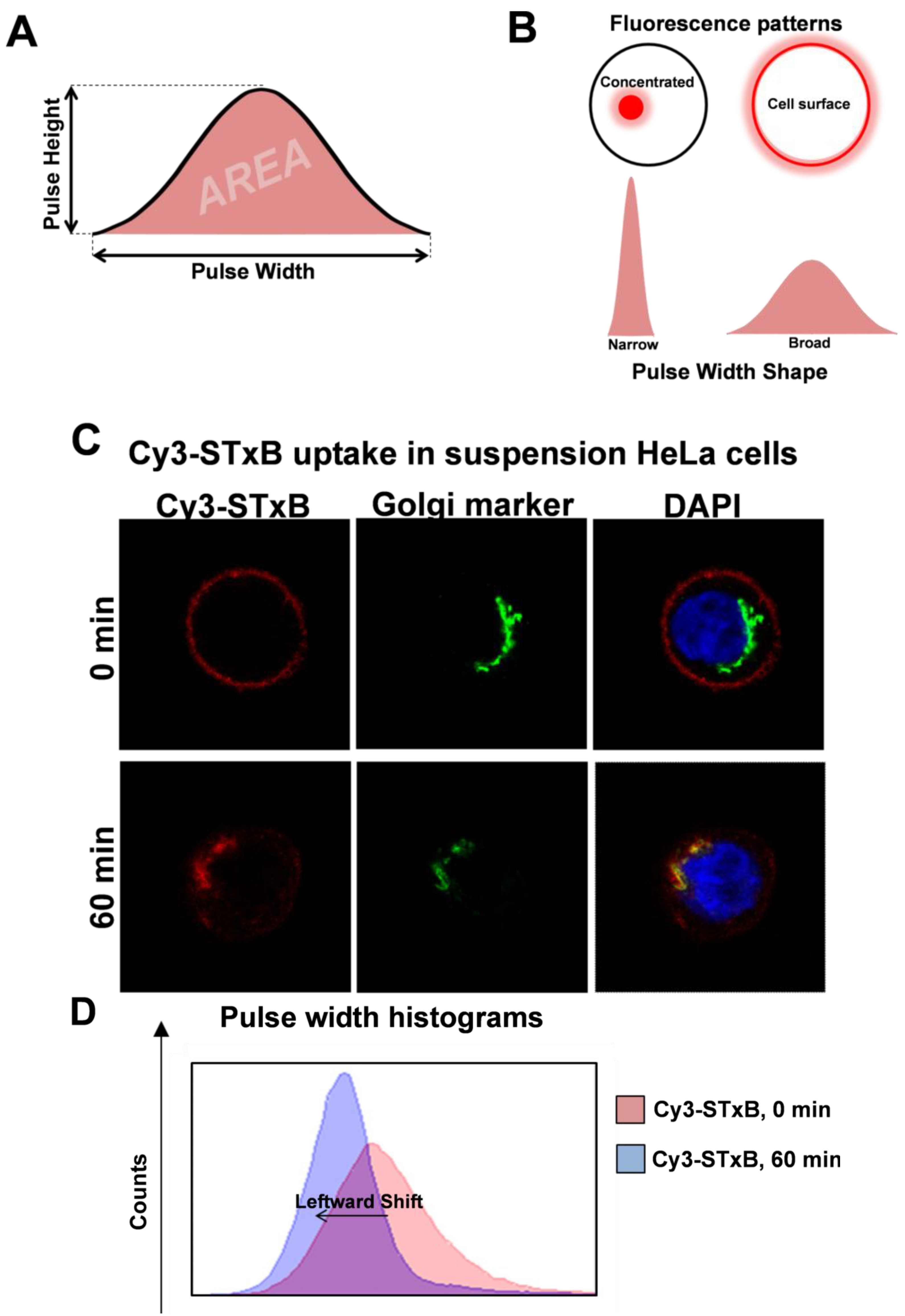
6. High Throughput Quantitation of Intracellular Events by Pulse Width Analysis
7. Concluding Remarks
Acknowledgements
References
- Gruenberg, J.; Griffiths, G.; Howell, K.E. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J. Cell Biol. 1989, 108, 1301–1316. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Johannes, L.; Popoff, V. Tracing the retrograde route in protein trafficking. Cell 2008, 135, 1175–1187. [Google Scholar] [CrossRef]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef]
- Ghosh, R.N.; Mallet, W.G.; Soe, T.T.; McGraw, T.E.; Maxfield, F.R. An endocytosed TGN38 chimeric protein is delivered to the TGN after trafficking through the endocytic recycling compartment in CHO cells. J. Cell Biol. 1998, 142, 923–936. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar] [CrossRef]
- Shewan, A.M.; van Dam, E.M.; Martin, S.; Luen, T.B.; Hong, W.; Bryant, N.J.; James, D.E. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: Involvement of an acidic targeting motif. Mol. Biol Cell 2003, 14, 973–986. [Google Scholar] [CrossRef]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef]
- Utskarpen, A.; Slagsvold, H.H.; Iversen, T.G.; Walchli, S.; Sandvig, K. Transport of ricin from endosomes to the Golgi apparatus is regulated by Rab6A and Rab6A'. Traffic 2006, 7, 663–672. [Google Scholar] [CrossRef]
- Abbe, E. Beitrage zur Theorie des Mikroskops und der mikroskopischen Wahrmehmung. Arch. Mikrosk. Anat. 1873, 9, 413–420. [Google Scholar] [CrossRef]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Patterson, G.H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009, 19, 555–565. [Google Scholar] [CrossRef]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell. Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef]
- Toomre, D.; Bewersdorf, J. A new wave of cellular imaging. Annu. Rev. Cell Dev. Biol. 2010, 26, 285–314. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar] [CrossRef]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar]
- Ach, T.; Best, G.; Rossberger, S.; Heintzmann, R.; Cremer, C.; Dithmar, S. Autofluorescence imaging of human RPE cell granules using structured illumination microscopy. Br. J. Ophthalmol. 2012, 96, 1141–1144. [Google Scholar] [CrossRef]
- Hanssen, E.; Carlton, P.; Deed, S.; Klonis, N.; Sedat, J.; DeRisi, J.; Tilley, L. Whole cell imaging reveals novel modular features of the exomembrane system of the malaria parasite, Plasmodium falciparum. Int J. Parasitol. 2010, 40, 123–134. [Google Scholar] [CrossRef]
- Meyer, L.; Wildanger, D.; Medda, R.; Punge, A.; Rizzoli, S.O.; Donnert, G.; Hell, S.W. Dual-color STED microscopy at 30-nm focal-plane resolution. Small 2008, 4, 1095–1100. [Google Scholar]
- Moneron, G.; Medda, R.; Hein, B.; Giske, A.; Westphal, V.; Hell, S.W. Fast STED microscopy with continuous wave fiber lasers. Opt. Express 2010, 18, 1302–1309. [Google Scholar]
- Schroder, J.; Benink, H.; Dyba, M.; Los, G.V. In vivo labeling method using a genetic construct for nanoscale resolution microscopy. Biophys. J. 2009, 96, L01–L03. [Google Scholar] [CrossRef]
- Rittweger, E.; Han, K.Y.; Irvine, S.E.; Eggeling, C.; Hell, S.W. STED microscopy reveals crystal colour centres with nanometric resolution. Nature Photonics 2009, 3, 144–147. [Google Scholar]
- Harke, B.; Ullal, C.K.; Keller, J.; Hell, S.W. Three-dimensional nanoscopy of colloidal crystals. Nano Lett. 2008, 8, 1309–1313. [Google Scholar]
- Hein, B.; Willig, K.I.; Hell, S.W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc. Natl. Acad. Sci USA 2008, 105, 14271–14276. [Google Scholar] [CrossRef]
- Donnert, G.; Keller, J.; Wurm, C.A.; Rizzoli, S.O.; Westphal, V.; Schonle, A.; Jahn, R.; Jakobs, S.; Eggeling, C.; Hell, S.W. Two-color far-field fluorescence nanoscopy. Biophys. J. 2007, 92, L67–L69. [Google Scholar] [CrossRef]
- Buckers, J.; Wildanger, D.; Vicidomini, G.; Kastrup, L.; Hell, S.W. Simultaneous multi-lifetime multi-color STED imaging for colocalization analyses. Opt. Express 2011, 19, 3130–3143. [Google Scholar]
- Schmidt, R.; Wurm, C.A.; Jakobs, S.; Engelhardt, J.; Egner, A.; Hell, S.W. Spherical nanosized focal spot unravels the interior of cells. Nat. Methods 2008, 5, 539–544. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef]
- Shtengel, G.; Galbraith, J.A.; Galbraith, C.G.; Lippincott-Schwartz, J.; Gillette, J.M.; Manley, S.; Sougrat, R.; Waterman, C.M.; Kanchanawong, P.; Davidson, M.W.; et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 3125–3130. [Google Scholar]
- Williamson, D.J.; Owen, D.M.; Rossy, J.; Magenau, A.; Wehrmann, M.; Gooding, J.J.; Gaus, K. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nat. Immunol. 2011, 12, 655–662. [Google Scholar] [CrossRef]
- Egner, A.; Geisler, C.; von Middendorff, C.; Bock, H.; Wenzel, D.; Medda, R.; Andresen, M.; Stiel, A.C.; Jakobs, S.; Eggeling, C.; et al. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys. J. 2007, 93, 3285–3290. [Google Scholar] [CrossRef]
- Bates, M.; Huang, B.; Dempsey, G.T.; Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007, 317, 1749–1753. [Google Scholar]
- Heilemann, M.; van de Linde, S.; Schuttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Edit. 2008, 47, 6172–6176. [Google Scholar]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar]
- Simmons, S.R.; Pawley, J.B.; Albrecht, R.M. Optimizing parameters for correlative immunogold localization by video-enhanced light microscopy, high-voltage transmission electron microscopy, and field emission scanning electron microscopy. J. Histochem. Cytochem. 1990, 38, 1781–1785. [Google Scholar] [CrossRef]
- Van Weering, J.R.; Brown, E.; Sharp, T.H.; Mantell, J.; Cullen, P.J.; Verkade, P. Intracellular membrane traffic at high resolution. Methods Cell Biol. 2010, 96, 619–648. [Google Scholar]
- Polishchuk, R.S.; Polishchuk, E.V.; Marra, P.; Alberti, S.; Buccione, R.; Luini, A.; Mironov, A.A. Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J. Cell Biol. 2000, 148, 45–58. [Google Scholar] [CrossRef]
- Polishchuk, R.S.; San Pietro, E.; Di Pentima, A.; Tete, S.; Bonifacino, J.S. Ultrastructure of long-range transport carriers moving from the trans Golgi network to peripheral endosomes. Traffic 2006, 7, 1092–1103. [Google Scholar] [CrossRef]
- Van Rijnsoever, C.; Oorschot, V.; Klumperman, J. Correlative light-electron microscopy (CLEM) combining live-cell imaging and immunolabeling of ultrathin cryosections. Nat. Methods 2008, 5, 973–980. [Google Scholar] [CrossRef]
- Van Weering, J.R.; Brown, E.; Sharp, T.H.; Mantell, J.; Cullen, P.J.; Verkade, P. Intracellular membrane traffic at high resolution. Methods Cell Biol. 2010, 96, 619–648. [Google Scholar]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar] [CrossRef]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 1999, 17, 969–973. [Google Scholar]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11984–11989. [Google Scholar] [CrossRef]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; Weissman, I.; Siebert, P. "Fluorescent timer": Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar]
- Bevis, B.J.; Glick, B.S. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat. Biotechnol. 2002, 20, 83–87. [Google Scholar]
- Gross, L.A.; Baird, G.S.; Hoffman, R.C.; Baldridge, K.K.; Tsien, R.Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci USA 2000, 97, 11990–11995. [Google Scholar]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Subach, F.V.; Subach, O.M.; Gundorov, I.S.; Morozova, K.S.; Piatkevich, K.D.; Cuervo, A.M.; Verkhusha, V.V. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat. Chem. Biol. 2009, 5, 118–126. [Google Scholar]
- Lippincott-Schwartz, J.; Altan-Bonnet, N.; Patterson, G.H. Photobleaching and photoactivation: Following protein dynamics in living cells. Nat. Cell. Biol. 2003, 5 (Suppl.), S7–S14. [Google Scholar]
- Lukyanov, K.A.; Chudakov, D.M.; Lukyanov, S.; Verkhusha, V.V. Innovation: Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 885–891. [Google Scholar]
- Remington, S.J. Fluorescent proteins: Maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006, 16, 714–721. [Google Scholar] [CrossRef]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131–133. [Google Scholar]
- Bates, M.; Blosser, T.R.; Zhuang, X. Short-range spectroscopic ruler based on a single-molecule optical switch. Phys. Rev. Lett. 2005. [Google Scholar] [CrossRef]
- Baddeley, D.; Jayasinghe, I.D.; Cremer, C.; Cannell, M.B.; Soeller, C. Light-induced dark states of organic fluochromes enable 30 nm resolution imaging in standard media. Biophys. J. 2009, 96, L22–L24. [Google Scholar]
- Dempsey, G.T.; Bates, M.; Kowtoniuk, W.E.; Liu, D.R.; Tsien, R.Y.; Zhuang, X. Photoswitching mechanism of cyanine dyes. J. Am. Chem. Soc. 2009, 131, 18192–18193. [Google Scholar]
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373. [Google Scholar]
- Habuchi, S.; Ando, R.; Dedecker, P.; Verheijen, W.; Mizuno, H.; Miyawaki, A.; Hofkens, J. Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proc. Natl. Acad. Sci. USA 2005, 102, 9511–9516. [Google Scholar]
- Fuchs, J.; Bohme, S.; Oswald, F.; Hedde, P.N.; Krause, M.; Wiedenmann, J.; Nienhaus, G.U. A photoactivatable marker protein for pulse-chase imaging with super-resolution. Nat. Methods 2010, 7, 627–630. [Google Scholar]
- Grabenbauer, M.; Geerts, W.J.; Fernadez-Rodriguez, J.; Hoenger, A.; Koster, A.J.; Nilsson, T. Correlative microscopy and electron tomography of GFP through photooxidation. Nat. Methods 2005, 2, 857–862. [Google Scholar]
- Shu, X.; Lev-Ram, V.; Deerinck, T.J.; Qi, Y.; Ramko, E.B.; Davidson, M.W.; Jin, Y.; Ellisman, M.H.; Tsien, R.Y. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011, 9, e1001041. [Google Scholar] [CrossRef]
- North, A.J. Seeing is believing? A beginners' guide to practical pitfalls in image acquisition. J. Cell Biol. 2006, 172, 9–18. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103, 857–862. [Google Scholar]
- Zinchuk, V.; Zinchuk, O.; Okada, T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem. Cytochem. 2007, 40, 101–111. [Google Scholar] [CrossRef]
- Bolte, S.; Talbot, C.; Boutte, Y.; Catrice, O.; Read, N.D.; Satiat-Jeunemaitre, B. FM-dyes as experimental probes for dissecting vesicle trafficking in living plant cells. J. Microsc. 2004, 214, 159–173. [Google Scholar]
- Roerdink, Jos B.T.M.; Meijster, A. The watershed transform: definitions, algorithms and parallelization strategies. Fund. Inform. 2000, 41, 187–226. [Google Scholar]
- Lachmanovich, E.; Shvartsman, D.E.; Malka, Y.; Botvin, C.; Henis, Y.I.; Weiss, A.M. Co-localization analysis of complex formation among membrane proteins by computerized fluorescence microscopy: Application to immunofluorescence co-patching studies. J. Microsc. 2003, 212, 122–131. [Google Scholar] [CrossRef]
- Boutte, Y.; Crosnier, M.T.; Carraro, N.; Traas, J.; Satiat-Jeunemaitre, B. The plasma membrane recycling pathway and cell polarity in plants: studies on PIN proteins. J. Cell Sci. 2006, 119, 1255–1265. [Google Scholar]
- Woodcroft, B.J.; Hammond, L.; Stow, J.L.; Hamilton, N.A. Automated organelle-based colocalization in whole-cell imaging. Cytometry A 2009, 75, 941–950. [Google Scholar]
- Jaskolski, F.; Mulle, C.; Manzoni, O.J. An automated method to quantify and visualize colocalized fluorescent signals. J. Neurosci. Methods 2005, 146, 42–49. [Google Scholar] [CrossRef]
- Hoffman, R.A. Unit 1 23 Pulse width for particle sizing. Curr. Protoc. Cytom. 2009. [Google Scholar] [CrossRef]
- Wersto, R.P.; Chrest, F.J.; Leary, J.F.; Morris, C.; Stetler-Stevenson, M.A.; Gabrielson, E. Doublet discrimination in DNA cell-cycle analysis. Cytometry 2001, 46, 296–306. [Google Scholar] [CrossRef]
- Ramdzan, Y.M.; Polling, S.; Chia, C.P.; Ng, I.H.; Ormsby, A.R.; Croft, N.P.; Purcell, A.W.; Bogoyevitch, M.A.; Ng, D.C.; Gleeson, P.A.; et al. Tracking protein aggregation and mislocalization in cells with flow cytometry. Nat. Methods 2012, 9, 467–470. [Google Scholar]
- Nakao, H.; Takeda, T. Escherichia coli Shiga toxin. J. Nat. Toxins 2000, 9, 299–313. [Google Scholar]
- Gruenberg, J. The endocytic pathway: A mosaic of domains. Nat. Rev. Mol. Cell Biol. 2001, 2, 721–730. [Google Scholar] [CrossRef]
- Lieu, Z.Z.; Gleeson, P.A. Endosome-to-Golgi transport pathways in physiological processes. Histol. Histopathol. 2011, 26, 395–408. [Google Scholar]
- Mari, M.; Bujny, M.V.; Zeuschner, D.; Geerts, W.J.; Griffith, J.; Petersen, C.M.; Cullen, P.J.; Klumperman, J.; Geuze, H.J. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 2008, 9, 380–393. [Google Scholar] [CrossRef]
- Kukulski, W.; Schorb, M.; Kaksonen, M.; Briggs, J.A. Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell 2012, 150, 508–520. [Google Scholar]
- Chan, W.C.; Nie, S. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 1998, 281, 2016–2018. [Google Scholar] [CrossRef]
- Fomenko, V.; Nesbitt, D.J. Solution control of radiative and nonradiative lifetimes: A novel contribution to quantum dot blinking suppression. Nano Lett. 2008, 8, 287–293. [Google Scholar] [CrossRef]
- Chien, C.Y.; Chang, Y.J.; Chen, K.H.; Lai, W.T.; George, T.; Scherer, A.; Li, P.W. Nanoscale, catalytically enhanced local oxidation of silicon-containing layers by 'burrowing' Ge quantum dots. Nanotechnology 2011. [Google Scholar] [CrossRef]
- Shi, X.; Xie, Z.; Song, Y.; Tan, Y.; Yeung, E.S.; Gai, H. Superlocalization spectral imaging microscopy of a multicolor quantum dot complex. Anal. Chem. 2012, 84, 1504–1509. [Google Scholar]
- Schieber, C.; Bestetti, A.; Lim, J.P.; Ryan, A.D.; Nguyen, T.L.; Eldridge, R.; White, A.R.; Gleeson, P.A.; Donnelly, P.S.; Williams, S.J.; Mulvaney, P. Conjugation of transferrin to azide-modified CdSe/ZnS core-shell quantum dots using cyclooctyne click chemistry. Angew. Chem. Int. Edit. 2012, 51, 10523–10527. [Google Scholar]
- Duong, T.; Goud, B.; Schauer, K. Closed-form density-based framework for automatic detection of cellular morphology changes. Proc. Natl. Acad. Sci. USA 2012, 109, 8382–8387. [Google Scholar] [CrossRef]
- Schauer, K.; Duong, T.; Bleakley, K.; Bardin, S.; Bornens, M.; Goud, B. Probabilistic density maps to study global endomembrane organization. Nat. Methods 2010, 7, 560–566. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chia, P.Z.C.; Gleeson, P.A. Imaging and Quantitation Techniques for Tracking Cargo along Endosome-to-Golgi Transport Pathways. Cells 2013, 2, 105-123. https://doi.org/10.3390/cells2010105
Chia PZC, Gleeson PA. Imaging and Quantitation Techniques for Tracking Cargo along Endosome-to-Golgi Transport Pathways. Cells. 2013; 2(1):105-123. https://doi.org/10.3390/cells2010105
Chicago/Turabian StyleChia, Pei Zhi Cheryl, and Paul A. Gleeson. 2013. "Imaging and Quantitation Techniques for Tracking Cargo along Endosome-to-Golgi Transport Pathways" Cells 2, no. 1: 105-123. https://doi.org/10.3390/cells2010105
APA StyleChia, P. Z. C., & Gleeson, P. A. (2013). Imaging and Quantitation Techniques for Tracking Cargo along Endosome-to-Golgi Transport Pathways. Cells, 2(1), 105-123. https://doi.org/10.3390/cells2010105