
TDGF1 Mediates the Oncogenic Effects of the OLMALINC/miR-3614-5p ceRNA Axis in Colon Cancer Through Nodal/Smad2 and Glypican-1/MAPK-AKT Signaling

 , ,
, ,
Highlights
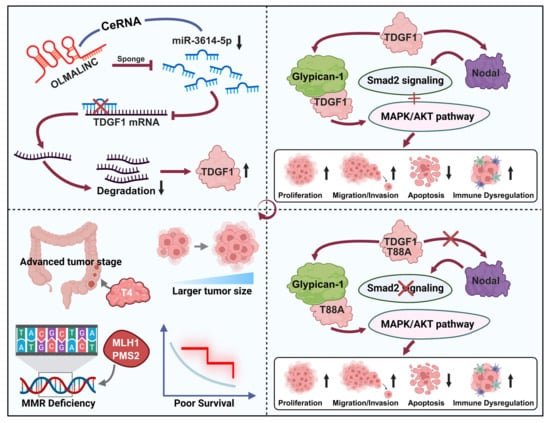
- OLMALINC sponges miR-3614-5p to upregulate TDGF1, driving colon cancer progression via Nodal/Smad2 and MAPK/AKT pathways.
- High TDGF1 expression correlates with an immunosuppressive microenvironment, advanced tumor stage, and poor prognosis.
- TDGF1 serves as a promising prognostic biomarker for predicting clinical outcomes in colon cancer patients.
- The OLMALINC/miR-3614-5p/TDGF1 regulatory axis provides potential targets for future therapeutic strategies.
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Database Analysis
2.2. GEPIA Database Analysis
2.3. TISIDB Database Analysis
2.4. miRNA Prediction Database Analysis
2.5. ENCORI Database Analysis
2.6. TIMER Database Analysis
2.7. LinkedOmics Database
2.8. STRING Database
2.9. CancerSCEM Database Analysis
2.10. Metascape Database Analysis
2.11. Cell Culture and Transfection
2.12. Luciferase Reporter Assay
2.13. Nuclear/Cytoplasmic Fractionation
2.14. Quantitative Real-Time PCR
2.15. Western Blot
2.16. Co-Immunoprecipitation Assay
2.17. MTT Assay
2.18. Colony Formation Assay
2.19. Transwell Assay
2.20. Flow Cytometry for Apoptosis Detection
2.21. In Vivo Tumorigenicity Assay
2.22. Immunohistochemistry (IHC)
2.23. Statistical Analysis
3. Results
3.1. TDGF1 Exhibits Distinct, Tumor-Specific Expression Patterns Independent of Pathological Stage
3.2. TDGF1 Expression Is Associated with Poor Disease-Free Survival in Colon Cancer
3.3. Hsa-miR-3614-5p Targets TDGF1 and Serves as a Favorable Prognostic Indicator in Colon Cancer
3.4. OLMALINC Functions as a Competing Endogenous RNA (ceRNA) by Targeting hsa-miR-3614-5p, and Its Elevated Expression Is Associated with Poor Outcome
3.5. OLMALINC Regulates TDGF1 via hsa-miR-3614-5p
3.6. The hsa-miR-3614-5p Overexpression or TDGF1 Suppression Reverses the Malignant Progression of Colon Cancer Driven by OLMALINC
3.7. Functional Rescue Assays in OLMALINC-Knockdown Cells Verify the hsa-miR-3614-5p/TDGF1 Axis as a Downstream Effector
3.8. TDGF1 Facilitates the Malignant Progression of Colon Cancer Cells, and Thr88 Mutation Partially Impairs Its Oncogenic Function
3.9. TDGF1 Mediates Dual Signaling Activation Through Thr88-Dependent Nodal/Smad2 and Glypican-1/MAPK-AKT Axes
3.10. TDGF1 Correlates with Immune Infiltration and Cell–Cell Communication, and Regulates Apoptosis in Colon Cancer
3.11. Knockdown of TDGF1 Suppresses Tumorigenesis in a Colon Cancer Xenograft Model
3.12. TDGF1 Is Established as a Prognostic Biomarker in Colon Cancer by Clinical Evidence, Linked to Advanced Tumor Stage and MMR Deficiency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TDGF1 | Teratocarcinoma-derived growth factor 1 |
| ceRNA | Competing endogenous RNA |
| COAD | Colon adenocarcinoma |
| EMT | Epithelial to mesenchymal transition |
| OS | Overall survival |
| DFS | Disease-free survival |
| ncRNAs | Noncoding RNAs |
| miRNAs | microRNAs |
| lncRNAs | Long noncoding RNAs |
| TIME | Tumor immune microenvironment |
| scRNA-seq | Single-cell RNA sequencing |
| TCGA | The Cancer Genome Atlas |
| TPM | Transcripts per million |
| ROC | Receiver operating characteristic |
| AUC | Area under the curve |
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| GO_BP | Gene Ontology biological process |
| GSEA | Gene set enrichment analysis |
| DEGs | Differentially expressed genes |
| PPI | Protein–protein interaction |
| wt | Wild-type |
| mut | Mutant |
| qRT-PCR | Quantitative real-time PCR |
| ECL | Enhanced chemiluminescence |
| IHC | Immunohistochemistry |
| SD | Standard deviation |
| HRs | Hazard ratios |
| CIs | Confidence intervals |
| sCNAs | Somatic copy number alterations |
| circRNAs | Circular RNAs |
References
- Cao, W.; Chen, H.D.; Yu, Y.W.; Li, N.; Chen, W.Q. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Thanikachalam, K.; Khan, G. Colorectal Cancer and Nutrition. Nutrients 2019, 11, 164. [Google Scholar] [CrossRef]
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.E.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Ciccodicola, A.; Dono, R.; Obici, S.; Simeone, A.; Zollo, M.; Persico, M.G. Molecular characterization of a gene of the ‘EGF family’ expressed in undifferentiated human NTERA2 teratocarcinoma cells. EMBO J. 1989, 8, 1987–1991. [Google Scholar] [CrossRef] [PubMed]
- Arnouk, H.; Yum, G.; Shah, D. Cripto-1 as a Key Factor in Tumor Progression, Epithelial to Mesenchymal Transition and Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 9280. [Google Scholar] [CrossRef] [PubMed]
- Saloman, D.S.; Bianco, C.; Ebert, A.D.; Khan, N.I.; De Santis, M.; Normanno, N.; Wechselberger, C.; Seno, M.; Williams, K.; Sanicola, M.; et al. The EGF-CFC family: Novel epidermal growth factor-related proteins in development and cancer. Endocr. Relat. Cancer 2000, 7, 199–226. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Maiello, M.R.; Carriero, M.V.; Rehman, A.; Wechselberger, C.; Arra, C.; Strizzi, L.; Sanicola, M.; et al. Cripto-1 overexpression leads to enhanced invasiveness and resistance to anoikis in human MCF-7 breast cancer cells. J. Cell. Physiol. 2004, 198, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Ebert, A.; Chang, C.; Rehman, A.; Normanno, N.; Guedez, L.; Salloum, R.; Ginsburg, E.; Sun, Y.; et al. Role of human cripto-1 in tumor angiogenesis. J. Natl. Cancer Inst. 2005, 97, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, B.; Gao, L.; Zhang, L.; Zhu, K.; Cheng, R.; Wang, C. Clinical significance of cripto-1 expression in lung adenocarcinoma. Oncotarget 2017, 8, 79087–79098. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yuan, Q.; Hu, H.; Wang, W.; Zhang, Q.; Li, L.; Wang, J.; Yang, R. Expression of Cripto-1 predicts poor prognosis in stage I non-small cell lung cancer. J. Cell. Mol. Med. 2020, 24, 9705–9711. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Y.; Zhang, L.H.; Jia, S.Q.; Shi, T.; Niu, Z.J.; Du, H.; Zhang, G.G.; Hu, Y.; Lu, A.P.; Li, J.Y.; et al. Positive association of up-regulated Cripto-1 and down-regulated E-cadherin with tumour progression and poor prognosis in gastric cancer. Histopathology 2008, 52, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Karasawa, H.; Suzuki, T.; Nakayama, S.; Katagiri, M.; Maeda, S.; Ohnuma, S.; Motoi, F.; Naitoh, T.; Unno, M. The Function and Prognostic Significance of Cripto-1 in Colorectal Cancer. Cancer Investig. 2020, 38, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cui, X.; Yu, X.; Bian, B.S.; Qian, F.; Hu, X.G.; Ji, C.D.; Yang, L.; Ren, Y.; Cui, W.; et al. Cripto-1 acts as a functional marker of cancer stem-like cells and predicts prognosis of the patients in esophageal squamous cell carcinoma. Mol. Cancer 2017, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Daraghma, H.; Untiveros, G.; Raskind, A.; Iaccarino, E.; Sandomenico, A.; Ruvo, M.; Arnouk, H.; Ciancio, M.J.; Cuevas-Nunez, M.; Strizzi, L. The role of Nodal and Cripto-1 in human oral squamous cell carcinoma. Oral Dis. 2021, 27, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Mancino, M.; Rehman, A.; Hamada, S.; Watanabe, K.; De Luca, A.; Jones, B.; Balogh, G.; Russo, J.; et al. Identification of cripto-1 as a novel serologic marker for breast and colon cancer. Clin. Cancer Res. 2006, 12, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H.; Chi, X.; Fan, Y.; Shi, Y.; Niu, J. High level of serum Cripto-1 in hepatocellular carcinoma, especially with hepatitis B virus infection. Medicine 2018, 97, e11781. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Wei, W.; Xu, J.; Guo, Z.X.; Xiao, C.Z.; Zhang, Y.F.; Jian, P.E.; Wu, X.L.; Shi, M.; Guo, R.P. Elevated expression of Cripto-1 correlates with poor prognosis in hepatocellular carcinoma. Oncotarget 2015, 6, 35116–35128. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.J.; Chen, S.N.; Chen, W.G.; Wu, G.Q.; Liao, Y.F.; Xu, J.B.; Tang, H.; Yang, S.H.; He, S.Y.; Luo, Y.F.; et al. Cripto-1 expression in patients with clear cell renal cell carcinoma is associated with poor disease outcome. J. Exp. Clin. Cancer Res. 2019, 38, 378. [Google Scholar] [CrossRef] [PubMed]
- Pattillo, R.A.; Hussa, R.O.; Story, M.T.; Ruckert, A.C.; Shalaby, M.R.; Mattingly, R.F. Tumor antigen and human chorionic gonadotropin in CaSki cells: A new epidermoid cervical cancer cell line. Science 1977, 196, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Yang, T.; Liu, R.; Xu, Y. Overexpression levels of cripto-1 predict poor prognosis in patients with prostate cancer following radical prostatectomy. Oncol. Lett. 2019, 18, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, Z.; Yang, K.; Liu, R.; Xu, Y. Cripto-1 promotes epithelial-mesenchymal transition in prostate cancer via Wnt/beta-catenin signaling. Oncol. Rep. 2017, 37, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Wechselberger, C.; Frank, S.; Wallace-Jones, B.; Seno, M.; Martinez-Lacaci, I.; Bianco, C.; De Santis, M.; Weitzel, H.K.; Salomon, D.S. Cripto-1 induces phosphatidylinositol 3′-kinase-dependent phosphorylation of AKT and glycogen synthase kinase 3beta in human cervical carcinoma cells. Cancer Res. 1999, 59, 4502–4505. [Google Scholar] [PubMed]
- D’Antonio, A.; Losito, S.; Pignata, S.; Grassi, M.; Perrone, F.; De Luca, A.; Tambaro, R.; Bianco, C.; Gullick, W.J.; Johnson, G.R.; et al. Transforming growth factor alpha, amphiregulin and cripto-1 are frequently expressed in advanced human ovarian carcinomas. Int. J. Oncol. 2002, 21, 941–948. [Google Scholar] [PubMed]
- De Luca, A.; Lamura, L.; Strizzi, L.; Roma, C.; D’Antonio, A.; Margaryan, N.; Pirozzi, G.; Hsu, M.Y.; Botti, G.; Mari, E.; et al. Expression and functional role of CRIPTO-1 in cutaneous melanoma. Br. J. Cancer 2011, 105, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N.; Sticht, C. miRWalk database for miRNA-target interactions. Methods Mol. Biol. 2014, 1182, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, Y.; Shang, Y.; Mai, J.; Shi, S.; Lu, M.; Bu, C.; Zhang, Z.; Zhang, Z.; Li, Y.; et al. CancerSCEM: A database of single-cell expression map across various human cancers. Nucleic Acids Res. 2022, 50, D1147–D1155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Adkins, H.B.; Wechselberger, C.; Seno, M.; Normanno, N.; De Luca, A.; Sun, Y.; Khan, N.; Kenney, N.; Ebert, A.; et al. Cripto-1 activates nodal- and ALK4-dependent and -independent signaling pathways in mammary epithelial Cells. Mol. Cell. Biol. 2002, 22, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Rehman, A.; Normanno, N.; Wechselberger, C.; Sun, Y.; Khan, N.; Hirota, M.; Adkins, H.; Williams, K.; et al. A Nodal- and ALK4-independent signaling pathway activated by Cripto-1 through Glypican-1 and c-Src. Cancer Res. 2003, 63, 1192–1197. [Google Scholar] [PubMed]
- Schiffer, S.G.; Foley, S.; Kaffashan, A.; Hronowski, X.; Zichittella, A.E.; Yeo, C.Y.; Miatkowski, K.; Adkins, H.B.; Damon, B.; Whitman, M.; et al. Fucosylation of Cripto is required for its ability to facilitate nodal signaling. J. Biol. Chem. 2001, 276, 37769–37778. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Ge, C.; Luo, Y.; Hou, X.; Haltiwanger, R.S.; Stanley, P. The threonine that carries fucose, but not fucose, is required for Cripto to facilitate Nodal signaling. J. Biol. Chem. 2007, 282, 20133–20141. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.T.; Liu, J.J.; Luo, Y.; E, C.; Haltiwanger, R.S.; Abate-Shen, C.; Shen, M.M. Dual roles of Cripto as a ligand and coreceptor in the nodal signaling pathway. Mol. Cell. Biol. 2002, 22, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.C.; Zhu, L.; Fan, Y.; Zhao, H.L. Clinicopathological and biological significance of cripto overexpression in human colon cancer. World J. Gastroenterol. 2013, 19, 8630–8637. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Zahra, M.H.; Takayanagi, A.; Seno, M. A Novel Artificially Humanized Anti-Cripto-1 Antibody Suppressing Cancer Cell Growth. Int. J. Mol. Sci. 2021, 22, 1709. [Google Scholar] [CrossRef] [PubMed]
- Razavi, Z.S.; Tajiknia, V.; Majidi, S.; Ghandali, M.; Mirzaei, H.R.; Rahimian, N.; Hamblin, M.R.; Mirzaei, H. Gynecologic cancers and non-coding RNAs: Epigenetic regulators with emerging roles. Crit. Rev. Oncol. Hematol. 2021, 157, 103192. [Google Scholar] [CrossRef] [PubMed]
- Lou, W.; Ding, B.; Wang, J.; Xu, Y. The Involvement of the hsa_circ_0088494-miR-876-3p-CTNNB1/CCND1 Axis in Carcinogenesis and Progression of Papillary Thyroid Carcinoma. Front. Cell Dev. Biol. 2020, 8, 605940. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Shoorei, H.; Anamag, F.T.; Taheri, M. The Role of Non-Coding RNAs in Controlling Cell Cycle Related Proteins in Cancer Cells. Front. Oncol. 2020, 10, 608975. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Ding, B.; Lou, W. microRNA-Dependent Modulation of Genes Contributes to ESR1’s Effect on ERalpha Positive Breast Cancer. Front. Oncol. 2020, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 Regulation by Noncoding RNAs in Cancers: The Present Knowledge and the Way Forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Shin, C.H.; Ji, H.; Jeong, S.D.; Park, M.S.; Won, H.H.; Pandey, P.R.; Tsitsipatis, D.; Gorospe, M.; Kim, H.H. hnRNPK-regulated LINC00263 promotes malignant phenotypes through miR-147a/CAPN2. Cell Death Dis. 2021, 12, 290. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Normanno, N.; Khan, N.; Salomon, D.S. Cripto-1: An oncofetal gene with many faces. Curr. Top. Dev. Biol. 2005, 67, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Waniczek, D.; Lorenc, Z.; Snietura, M.; Wesecki, M.; Kopec, A.; Muc-Wierzgon, M. Tumor-Associated Macrophages and Regulatory T Cells Infiltration and the Clinical Outcome in Colorectal Cancer. Arch. Immunol. Ther. Exp. 2017, 65, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, L.; Li, Y.; Long, Y.; Zhao, Q.; Ouyang, Y.; Bao, W.; Gong, K. Tumor-associated macrophage infiltration and prognosis in colorectal cancer: Systematic review and meta-analysis. Int. J. Color. Dis. 2020, 35, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. The Role of the Immune Infiltrate in Distinct Cancer Types and Its Clinical Implications: Lymphocytic Infiltration in Colorectal Cancer. Cancer Treat. Res. 2020, 180, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Bao, Y.L.; Yu, C.L.; Wang, Y.M.; Song, Z.B. Cripto-1 modulates macrophage cytokine secretion and phagocytic activity via NF-kappaB signaling. Immunol. Res. 2016, 64, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Xiao, W.; Hu, H.; Lu, G.; Chen, L.; Sun, Z.; Lu, M.; Ma, W.; Jiang, T.; Gao, Y.; et al. Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer. Clin. Transl. Med. 2021, 11, e422. [Google Scholar] [CrossRef] [PubMed]
- Alowaidi, F.; Hashimi, S.M.; Nguyen, M.; Meshram, M.; Alqurashi, N.; Cavanagh, B.L.; Bellette, B.; Ivanovski, S.; Meedenyia, A.; Wood, S.A. Investigating the role of CRIPTO-1 (TDGF-1) in glioblastoma multiforme U87 cell line. J. Cell. Biochem. 2018, 120, 7412–7427. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.; Kilic, M.; Tatli Dogan, H. The Relationship Between DNA Mismatch Repair Status and Clinicopathologic Characteristics in Colon Cancer. Turk. J. Gastroenterol. 2024, 35, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Bian, Y.; Lin, Y.; Li, S.; Jiang, L.; Wu, J.; Xie, Y.; Ning, S.; Liu, Y.; Huang, J.; et al. Human mesenchymal stem cell-derived miR-3614-5p exosomes suppress progression of colorectal cancer by targeting IL7Ralpha. J. Transl. Med. 2026. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Cell | Biomarker | R Value | Adjusted p Value |
|---|---|---|---|
| B cell | CD19 | −0.14 a | 2.72 × 10−2 * |
| CD79A | −0.15 a | 1.97 × 10−2 * | |
| CD8+ T cell | CD8A | −0.28 a | 2.52 × 10−5 ** |
| CD8B | −0.12 | 6.07 × 10−2 | |
| CD4+ T cell | CD4 | −0.22 a | 6.30 × 10−4 ** |
| M1 macrophage | NOS2 | −0.17 a | 5.46 × 10−3 ** |
| IRF5 | −0.017 | 7.80 × 10−1 | |
| PTGS2 | −0.19 a | 3.00 × 10−3 ** | |
| M2 macrophage | CD163 | −0.2 a | 1.41 × 10−3 ** |
| VSIG4 | −0.22 a | 7.47 × 10−4 ** | |
| MS4A4A | −0.24 a | 2.60 × 10−4 ** | |
| Neutrophil | CEACAM8 | −0.034 | 6.09 × 10−1 |
| ITGAM | −0.19 a | 2.91 × 10−3 ** | |
| CCR7 | −0.2 a | 1.75 × 10−3 ** | |
| Dendritic cell | HLA-DPB1 | −0.26 a | 5.09 × 10−5 ** |
| HLA-DQB1 | −0.22 a | 7.47 × 10−4 ** | |
| HLA-DRA | −0.32 a | 1.87 × 10−6 ** | |
| HLA-DPA1 | −0.28 a | 2.52 × 10−5 ** | |
| CD1C | −0.1 | 1.07 × 10−1 | |
| NRP1 | −0.2 a | 1.75 × 10−3 ** | |
| ITGAX | −0.22 a | 6.30 × 10−4 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Gao, F.; Li, X.; Li, J.; Yang, S.; Zhang, B.; Sun, Y.; Zheng, L.; Wang, G.; Liu, L.; Bao, Y.; et al. TDGF1 Mediates the Oncogenic Effects of the OLMALINC/miR-3614-5p ceRNA Axis in Colon Cancer Through Nodal/Smad2 and Glypican-1/MAPK-AKT Signaling. Cells 2026, 15, 1141. https://doi.org/10.3390/cells15131141
Gao F, Li X, Li J, Yang S, Zhang B, Sun Y, Zheng L, Wang G, Liu L, Bao Y, et al. TDGF1 Mediates the Oncogenic Effects of the OLMALINC/miR-3614-5p ceRNA Axis in Colon Cancer Through Nodal/Smad2 and Glypican-1/MAPK-AKT Signaling. Cells. 2026; 15(13):1141. https://doi.org/10.3390/cells15131141
Chicago/Turabian StyleGao, Feng, Xiaoli Li, Jiawei Li, Shuo Yang, Boyu Zhang, Ying Sun, Lihua Zheng, Guannan Wang, Lei Liu, Yongli Bao, and et al. 2026. "TDGF1 Mediates the Oncogenic Effects of the OLMALINC/miR-3614-5p ceRNA Axis in Colon Cancer Through Nodal/Smad2 and Glypican-1/MAPK-AKT Signaling" Cells 15, no. 13: 1141. https://doi.org/10.3390/cells15131141
APA StyleGao, F., Li, X., Li, J., Yang, S., Zhang, B., Sun, Y., Zheng, L., Wang, G., Liu, L., Bao, Y., & Yang, X. (2026). TDGF1 Mediates the Oncogenic Effects of the OLMALINC/miR-3614-5p ceRNA Axis in Colon Cancer Through Nodal/Smad2 and Glypican-1/MAPK-AKT Signaling. Cells, 15(13), 1141. https://doi.org/10.3390/cells15131141