

The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis



Abstract
1. Introduction
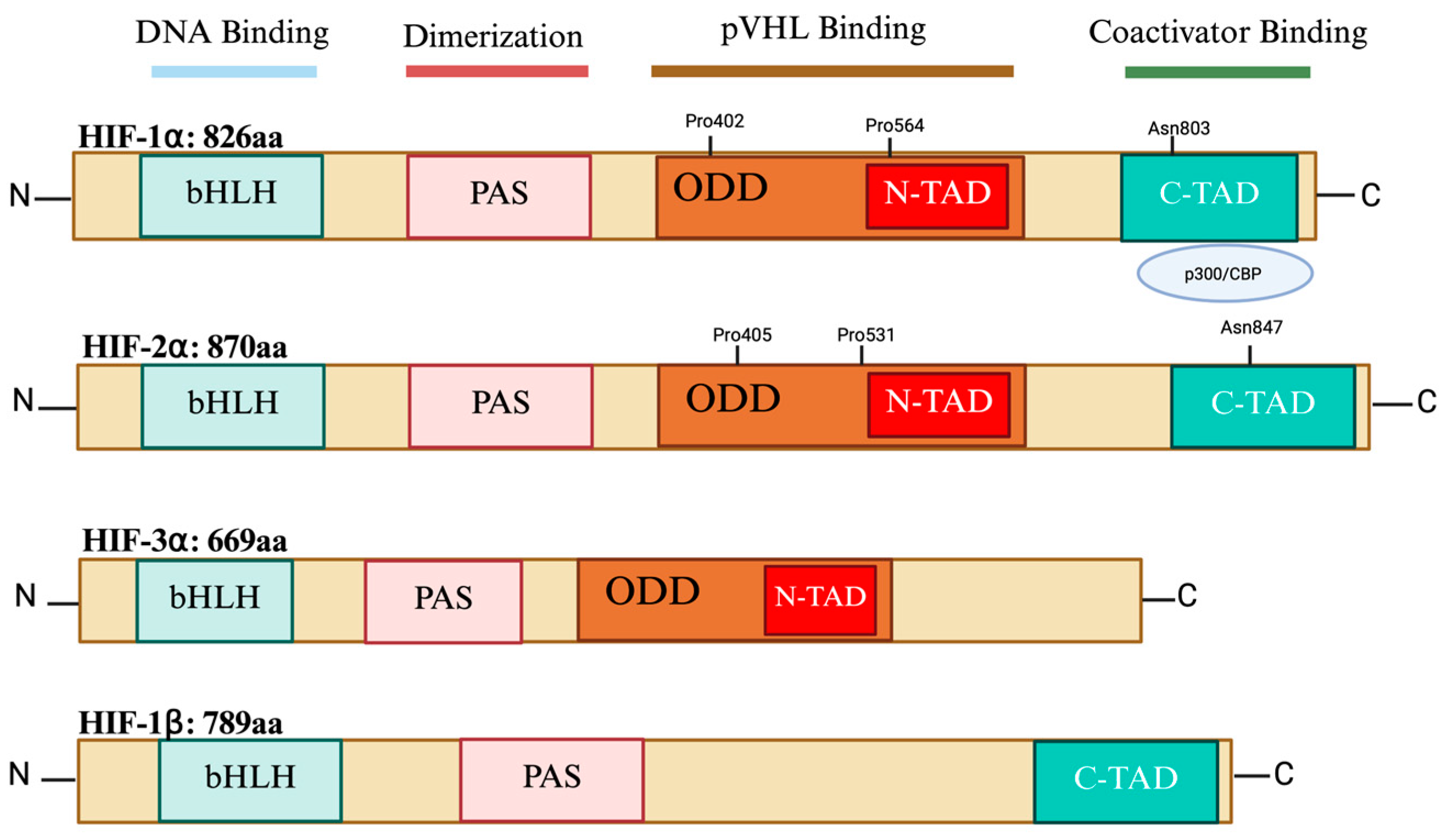
2. HIF Isoforms’ Structure
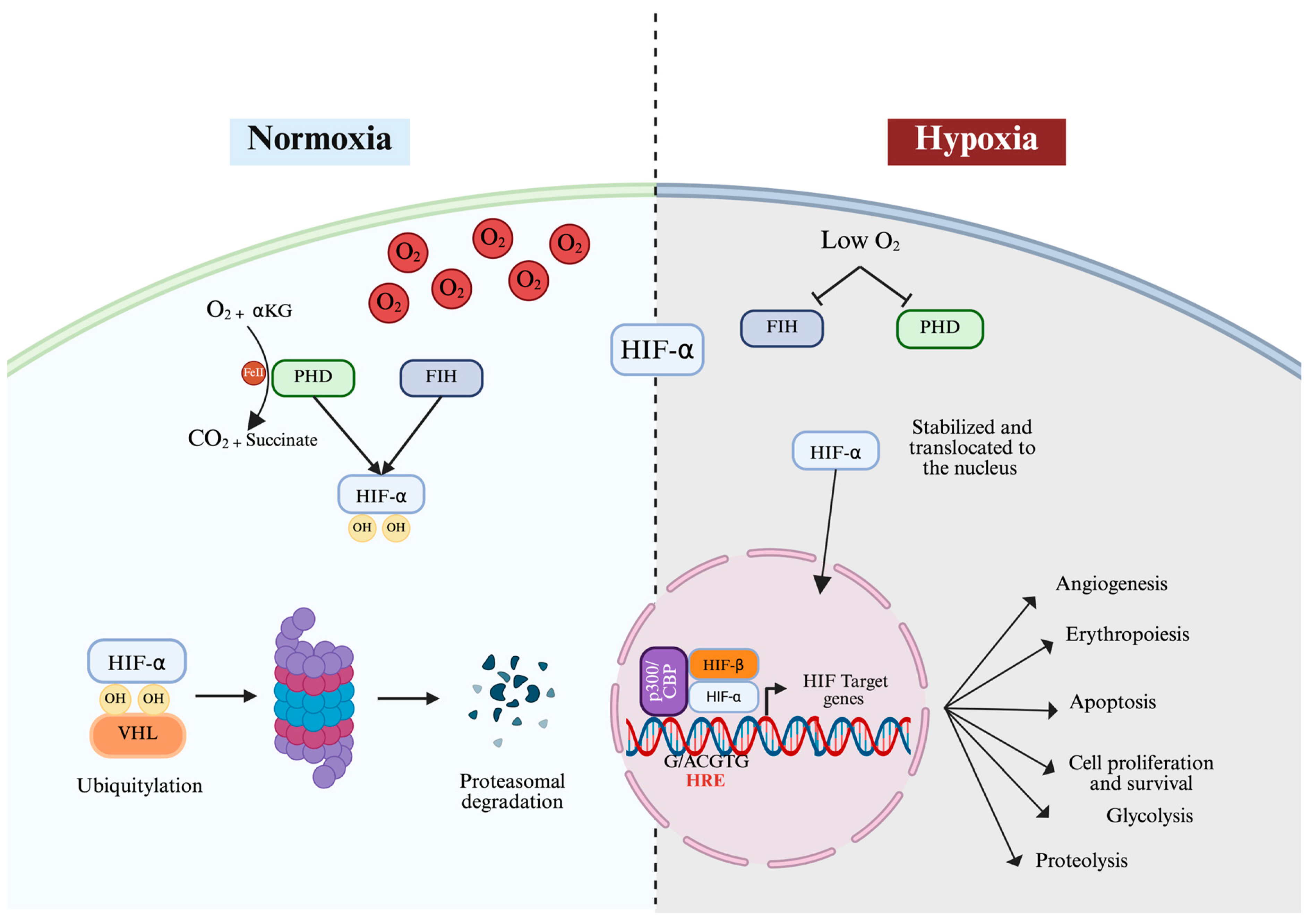
3. Oxygen-Dependent Regulation of HIF
4. Oxygen-Independent Regulation of HIF
5. Differential Expression Patterns and Functions of HIF-1α and HIF-2α
6. The Differential Role of HIF-1α and HIF-2α in the Metabolic Switch from Oxidative Phosphorylation to Glycolysis
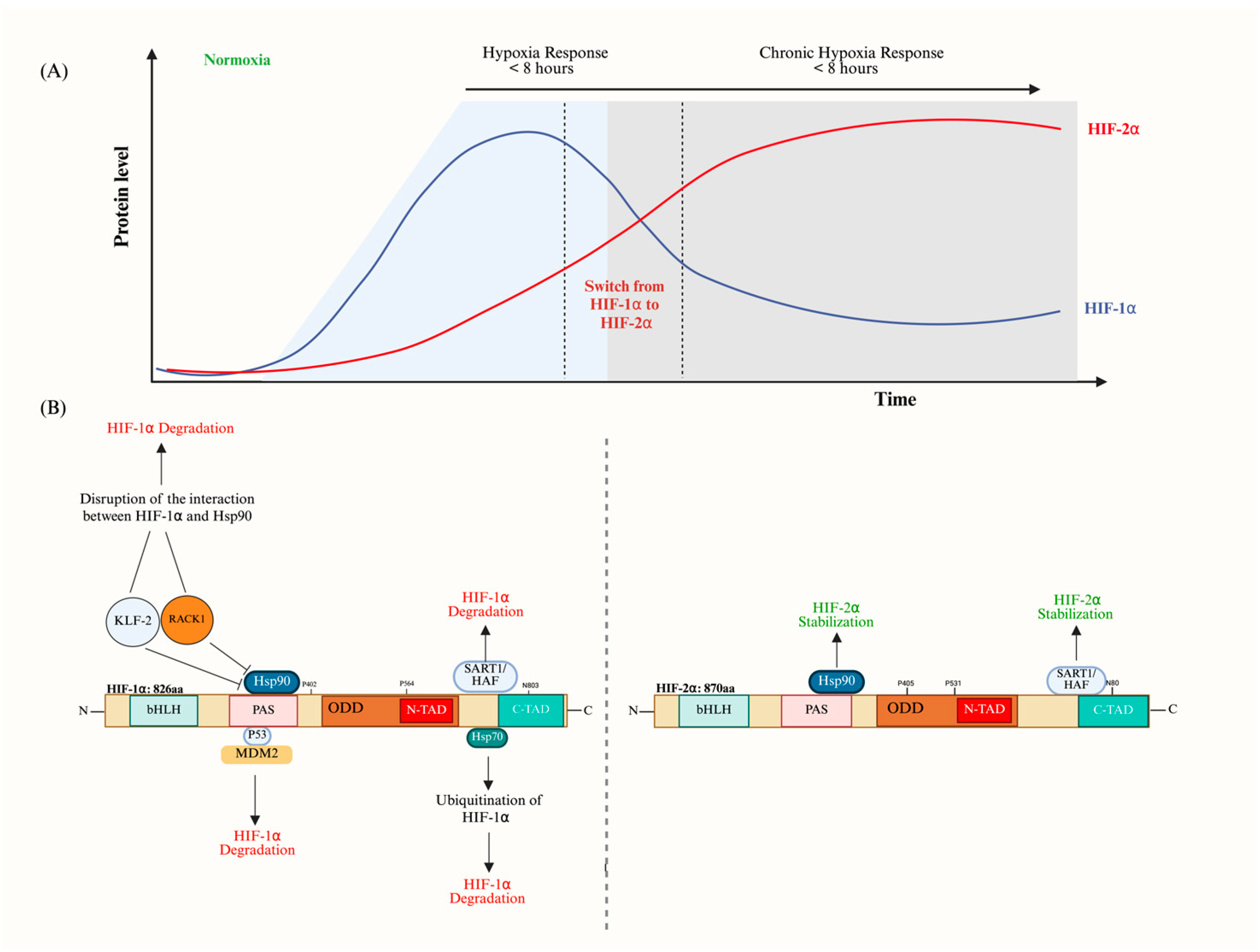
7. The Interrelation Between HIF-1α and HIF-2α (The HIF Switch)
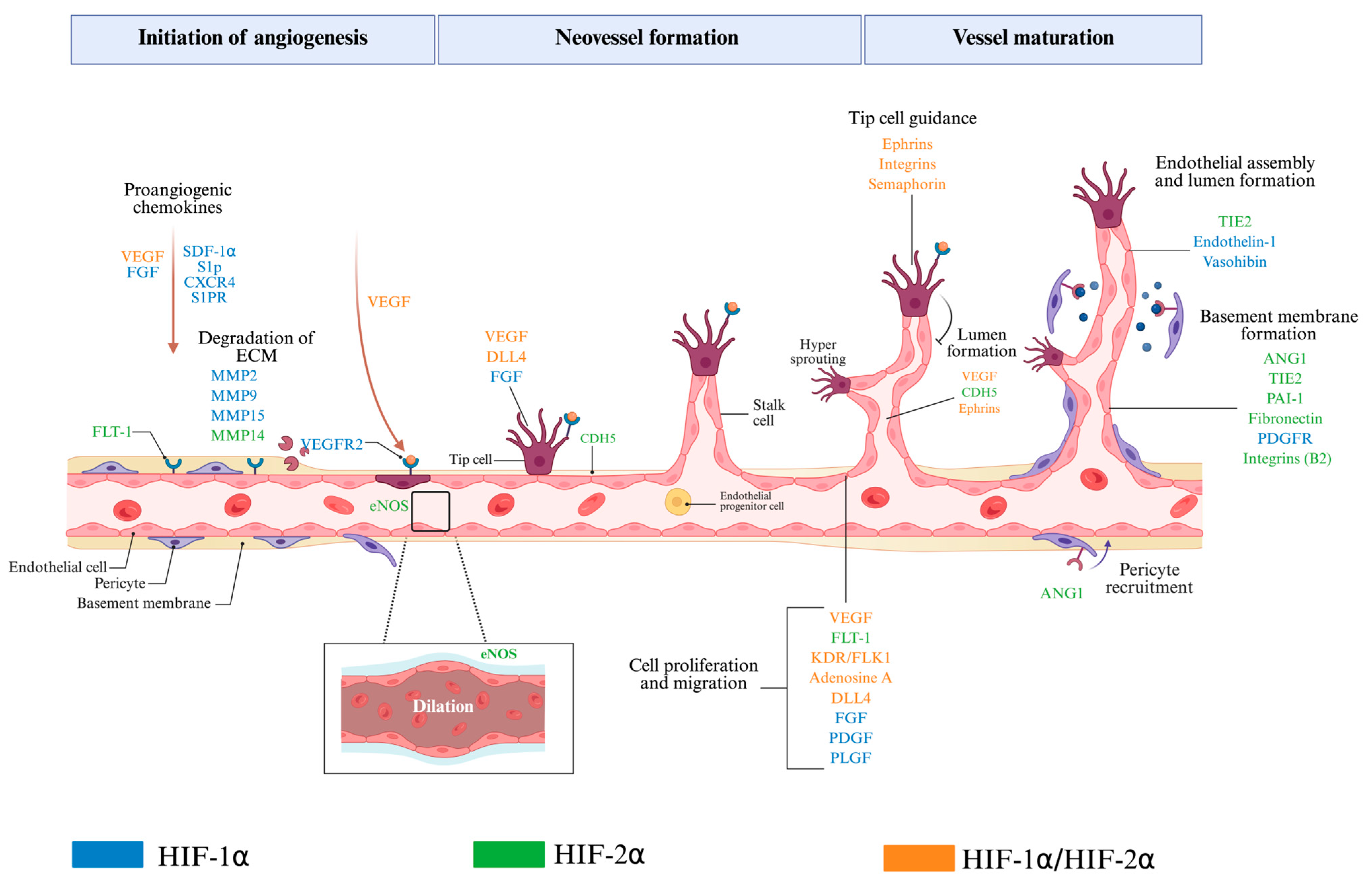
8. Role of HIF-1α and HIF-2α in Angiogenesis
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen Availability and Metabolic Adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Life with Oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C. The Hypoxia Response Pathways—Hats Off! N. Engl. J. Med. 2016, 375, 1687–1689. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 Transcription Factors—Similar but Not Identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Florczyk, U.; Czauderna, S.; Stachurska, A.; Tertil, M.; Nowak, W.; Kozakowska, M.; Poellinger, L.; Jozkowicz, A.; Loboda, A.; Dulak, J. Opposite Effects of HIF-1α and HIF-2α on the Regulation of IL-8 Expression in Endothelial Cells. Free Radic. Biol. Med. 2011, 51, 1882–1892. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibasaki, F. Hypoxia-Inducible Factor as an Angiogenic Master Switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular 02 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. New Horizons in Hypoxia Signaling Pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, H.; Li, H.; Ge, J. Molecular Basis and Clinical Implications of HIFs in Cardiovascular Diseases. Trends Mol. Med. 2022, 28, 916–938. [Google Scholar] [CrossRef] [PubMed]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The Mammalian Basic Helix–Loop–Helix/PAS Family of Transcriptional Regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Erbel, P.J.A.; Card, P.B.; Karakuzu, O.; Bruick, R.K.; Gardner, K.H. Structural Basis for PAS Domain Heterodimerization in the Basic Helix–Loop–Helix-PAS Transcription Factor Hypoxia-Inducible Factor. Proc. Natl. Acad. Sci. USA 2003, 100, 15504–15509. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of Oxygen Signaling at the Consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural Integration in Hypoxia-Inducible Factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the Baton: The HIF Switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers 2022, 14, 1259. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-Inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Metzen, E.; Stiehl, D.P.; Doege, K.; Marxsen, J.H.; Hellwig-Bürgel, T.; Jelkmann, W. Regulation of the Prolyl Hydroxylase Domain Protein 2 (Phd2 / Egln-1) Gene: Identification of a Functional Hypoxia-Responsive Element. Biochem. J. 2005, 387, 711–717. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling Rivalry in Hypoxic Tumor Growth and Progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef]
- Strowitzki, M.; Cummins, E.; Taylor, C. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef]
- Wong, B.W.; Kuchnio, A.; Bruning, U.; Carmeliet, P. Emerging Novel Functions of the Oxygen-Sensing Prolyl Hydroxylase Domain Enzymes. Trends Biochem. Sci. 2013, 38, 3–11. [Google Scholar] [CrossRef]
- Metzen, E.; Berchner-Pfannschmidt, U.; Stengel, P.; Marxsen, J.H.; Stolze, I.; Klinger, M.; Huang, W.Q.; Wotzlaw, C.; Hellwig-Bürgel, T.; Jelkmann, W.; et al. Intracellular Localisation of Human HIF-1α Hydroxylases:Implications for Oxygen Sensing. J. Cell Sci. 2003, 116, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Tal, R.; Shaish, A.; Bangio, L.; Peled, M.; Breitbart, E.; Harats, D. Activation of C-Transactivation Domain Is Essential for Optimal HIF-1α-Mediated Transcriptional and Angiogenic Effects. Microvasc. Res. 2008, 76, 1–6. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes. Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Singleton, R.S.; Sekirnik, R.; Trudgian, D.C.; Ambrose, L.J.; Miranda, M.X.; Tian, Y.; Kessler, B.M.; Schofield, C.J.; Ratcliffe, P.J. The FIH Hydroxylase Is a Cellular Peroxide Sensor That Modulates HIF Transcriptional Activity. EMBO Rep. 2012, 13, 251–257. [Google Scholar] [CrossRef]
- Cavadas, M.A.; Nguyen, L.K.; Cheong, A. Hypoxia-Inducible Factor (HIF) Network: Insights from Mathematical Models. Cell Commun. Signal 2013, 11, 42. [Google Scholar] [CrossRef]
- Minervini, G.; Pennuto, M.; Tosatto, S.C.E. The pVHL Neglected Functions, a Tale of Hypoxia-Dependent and -Independent Regulations in Cancer. Open Biol. 2020, 10, 200109. [Google Scholar] [CrossRef]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef]
- Daly, L.A.; Brownridge, P.J.; Batie, M.; Rocha, S.; Sée, V.; Eyers, C.E. Oxygen-Dependent Changes in Binding Partners and Post-Translational Modifications Regulate the Abundance and Activity of HIF-1α/2α. Sci. Signal. 2021, 14, eabf6685. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Yatsula, B.; Maier, C.L.; Sadler, T.J.; Whittaker, L.W.; Pober, J.S. Interferon-Gamma Induces Prolyl Hydroxylase (PHD)3 through a STAT1-Dependent Mechanism in Human Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1363–1369. [Google Scholar] [CrossRef]
- Qiu, B.; Yuan, P.; Du, X.; Jin, H.; Du, J.; Huang, Y. Hypoxia Inducible Factor-1α Is an Important Regulator of Macrophage Biology. Heliyon 2023, 9, e17167. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Malkov, M.I.; Lee, C.T.; Taylor, C.T. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 2021, 10, 2340. [Google Scholar] [CrossRef]
- Larsen, H.; Muz, B.; Khong, T.L.; Feldmann, M.; Paleolog, E.M. Differential Effects of Th1 versus Th2 Cytokines in Combination with Hypoxia on HIFs and Angiogenesis in RA. Arthritis Res. Ther. 2012, 14, R180. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic Induction of the Hypoxia-inducible Factor 1α by Insulin and Interleukin-1β Involves the Phosphatidylinositol 3-kinase Pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Lee, A.R.; Choi, J.W.; Lee, C.R.; Cho, K.-H.; Lee, J.H.; Cho, M.-L. IL-17 Induces Autophagy Dysfunction to Promote Inflammatory Cell Death and Fibrosis in Keloid Fibroblasts via the STAT3 and HIF-1α Dependent Signaling Pathways. Front. Immunol. 2022, 13, 888719. [Google Scholar] [CrossRef]
- Gai, X.; Zhou, P.; Xu, M.; Liu, Z.; Zheng, X.; Liu, Q. Hyperactivation of IL-6/STAT3 Pathway Leaded to the Poor Prognosis of Post-TACE HCCs by HIF-1α/SNAI1 Axis-Induced Epithelial to Mesenchymal Transition. J. Cancer 2020, 11, 570–582. [Google Scholar] [CrossRef]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential Activation and Antagonistic Function of HIF-a Isoforms in Macrophages Are Essential for NO Homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef]
- Mallikarjuna, P.; Raviprakash, T.S.; Aripaka, K.; Ljungberg, B.; Landström, M. Interactions between TGF-β Type I Receptor and Hypoxia-Inducible Factor-α Mediates a Synergistic Crosstalk Leading to Poor Prognosis for Patients with Clear Cell Renal Cell Carcinoma. Cell Cycle 2019, 18, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Bakker, W.J.; Harris, I.S.; Mak, T.W. FOXO3a Is Activated in Response to Hypoxic Stress and Inhibits HIF1-Induced Apoptosis via Regulation of CITED2. Mol. Cell 2007, 28, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate: Regulation of Hypoxia-Inducible Factor-1a. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Wu, C.-A.; Huang, D.-Y.; Lin, W.-W. Beclin-1-Independent Autophagy Positively Regulates Internal Ribosomal Entry Site-Dependent Translation of Hypoxia-Inducible Factor 1α under Nutrient Deprivation. Oncotarget 2014, 5, 7525–7539. [Google Scholar] [CrossRef]
- Nishimoto, A.; Kugimiya, N.; Hosoyama, T.; Enoki, T.; Li, T.-S.; Hamano, K. HIF-1α Activation under Glucose Deprivation Plays a Central Role in the Acquisition of Anti-Apoptosis in Human Colon Cancer Cells. Int. J. Oncol. 2014, 44, 2077–2084. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate Activates HIF-1 in Oxidative but Not in Warburg-Phenotype Human Tumor Cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [PubMed]
- Kappler, M.; Pabst, U.; Rot, S.; Taubert, H.; Wichmann, H.; Schubert, J.; Bache, M.; Weinholdt, C.; Immel, U.-D.; Grosse, I.; et al. Normoxic Accumulation of HIF1α Is Associated with Glutaminolysis. Clin. Oral Investig. 2017, 21, 211–224. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 Drives HIF-1α and VEGF-A Signalling via Multiple Mechanisms Involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef]
- Nayak, B.K.; Feliers, D.; Sudarshan, S.; Friedrichs, W.E.; Day, R.T.; New, D.D.; Fitzgerald, J.P.; Eid, A.; DeNapoli, T.; Parekh, D.J.; et al. Stabilization of HIF-2α through Redox Regulation of mTORC2 Activation and Initiation of mRNA Translation. Oncogene 2013, 32, 3147–3155. [Google Scholar] [CrossRef]
- Befani, C.; Liakos, P. The Role of Hypoxia-inducible Factor-2 Alpha in Angiogenesis. J. Cell. Physiol. 2018, 233, 9087–9098. [Google Scholar] [CrossRef]
- Aventaggiato, M.; Barreca, F.; Sansone, L.; Pellegrini, L.; Russo, M.A.; Cordani, M.; Tafani, M. Sirtuins and Hypoxia in EMT Control. Pharmaceuticals 2022, 15, 737. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Park, J.-H.; Choi, H.-J.; Won, H.-Y.; Joo, H.; Shin, D.-H.; Park, M.K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 Demethylates HIF1α to Inhibit Hydroxylation and Ubiquitin-Mediated Degradation in Tumor Angiogenesis. Oncogene 2017, 36, 5512–5521. [Google Scholar] [CrossRef]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the Histone Demethylase JMJD1A by Hypoxia-Inducible Factor 1α Enhances Hypoxic Gene Expression and Tumor Growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef]
- Jeong, J.-W.; Bae, M.-K.; Ahn, M.-Y.; Kim, S.-H.; Sohn, T.-K.; Bae, M.-H.; Yoo, M.-A.; Song, E.J.; Lee, K.-J.; Kim, K.-W. Regulation and Destabilization of HIF-1α by ARD1-Mediated Acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Xenaki, G.; Ontikatze, T.; Rajendran, R.; Stratford, I.J.; Dive, C.; Krstic-Demonacos, M.; Demonacos, C. PCAF Is an HIF-1α Cofactor That Regulates P53 Transcriptional Activity in Hypoxia. Oncogene 2008, 27, 5785–5796. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Lafleur, V.N.; Richard, S.; Richard, D.E. Transcriptional Repression of Hypoxia-Inducible Factor-1 (HIF-1) by the Protein Arginine Methyltransferase PRMT1. MBoC 2014, 25, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiao, W. Repression of Hypoxia-Inducible Factor α Signaling by Set7-Mediated Methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef]
- Lim, J.-H.; Choi, Y.-J.; Cho, C.-H.; Park, J.-W. Protein Arginine Methyltransferase 5 Is an Essential Component of the Hypoxia-Inducible Factor 1 Signaling Pathway. Biochem. Biophys. Res. Commun. 2012, 418, 254–259. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Lachance, G.; Uniacke, J.; Audas, T.E.; Holterman, C.E.; Franovic, A.; Payette, J.; Lee, S. DNMT3a Epigenetic Program Regulates the HIF-2α Oxygen-Sensing Pathway and the Cellular Response to Hypoxia. Proc. Natl. Acad. Sci. USA 2014, 111, 7783–7788. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential Gene Up-Regulation by Hypoxia-Inducible Factor-1A and Hypoxia-Inducible Factor-2A in HEK293T Cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1a (HIF-1a) and HIF-2a in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The Transcriptional Factors HIF-1 and HIF-2 and Their Novel Inhibitors in Cancer Therapy. Expert. Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 versus HIF-2—Is One More Important than the Other? Vasc. Pharmacol. 2012, 56, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef]
- Richter, S.; Qin, N.; Pacak, K.; Eisenhofer, G. Role of Hypoxia and HIF2α in Development of the Sympathoadrenal Cell Lineage and Chromaffin Cell Tumors with Distinct Catecholamine Phenotypic Features. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 68, pp. 285–317. ISBN 978-0-12-411512-5. [Google Scholar]
- Krishnan, J.; Ahuja, P.; Bodenmann, S.; Knapik, D.; Perriard, E.; Krek, W.; Perriard, J.-C. Essential Role of Developmentally Activated Hypoxia-Inducible Factor 1α for Cardiac Morphogenesis and Function. Circ. Res. 2008, 103, 1139–1146. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to Common Target Genes Is Differentially Regulated in Neuroblastoma: HIF-2α Promotes an Aggressive Phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Cowman, S.J.; Koh, M.Y. Revisiting the HIF Switch in the Tumor and Its Immune Microenvironment. Trends Cancer 2022, 8, 28–42. [Google Scholar] [CrossRef]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding Specificities of the HIF-1α and HIF-2α Transcription Factors in Chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-Resolution Genome-Wide Mapping of HIF-Binding Sites by ChIP-Seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-Inducible Factors HIF-1a and HIF-2-a. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 Mediates Hypoxia-Inducible HIF1α Activation in Kidney Tubular Epithelial Cells. Am. J. Physiol.—Ren. Physiol. 2021, 320, F464–F474. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, A.; Cornelissen, P.W.; Kirchmaier, B.C.; Mokry, M.; Iich, E.; Nirmala, E.; Liang, K.-H.; D. Végh, A.M.; Scholman, K.T.; Groot Koerkamp, M.J.; et al. Genome-Wide Analysis Reveals NRP1 as a Direct HIF1α-E2F7 Target in the Regulation of Motorneuron Guidance in Vivo. Nucleic Acids Res. 2016, 44, 3549–3566. [Google Scholar] [CrossRef]
- Soehngen, E.; Schaefer, A.; Koeritzer, J.; Huelsmeyer, V.; Zimmer, C.; Ringel, F.; Gempt, J.; Schlegel, J. Hypoxia Upregulates Aldehyde Dehydrogenase Isoform 1 (ALDH1) Expression and Induces Functional Stem Cell Characteristics in Human Glioblastoma Cells. Brain Tumor Pathol. 2014, 31, 247–256. [Google Scholar] [CrossRef]
- Sowa, T.; Menju, T.; Chen-Yoshikawa, T.F.; Takahashi, K.; Nishikawa, S.; Nakanishi, T.; Shikuma, K.; Motoyama, H.; Hijiya, K.; Aoyama, A.; et al. Hypoxia-Inducible Factor 1 Promotes Chemoresistance of Lung Cancer by Inducing Carbonic Anhydrase IX Expression. Cancer Med. 2017, 6, 288–297. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1*. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Jean, J.-C.; Rich, C.B.; Joyce-Brady, M. Hypoxia Results in an HIF-1-Dependent Induction of Brain-Specific Aldolase C in Lung Epithelial Cells. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2006, 291, L950–L956. [Google Scholar] [CrossRef]
- Gess, B.; Hofbauer, K.-H.; Deutzmann, R.; Kurtz, A. Hypoxia Up-Regulates Triosephosphate Isomerase Expression via a HIF-Dependent Pathway. Pflügers Arch. 2004, 448, 175–180. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 Mediates Metabolic Responses to Intratumoral Hypoxia and Oncogenic Mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Jung, F.; Palmer, L.A.; Zhou, N.; Johns, R.A. Hypoxic Regulation of Inducible Nitric Oxide Synthase via Hypoxia Inducible Factor-1 in Cardiac Myocytes. Circ. Res. 2000, 86, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia Induces the Expression of the Pro-Apoptotic Gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef]
- Guan, G.; Zhang, Y.; Lu, Y.; Liu, L.; Shi, D.; Wen, Y.; Yang, L.; Ma, Q.; Liu, T.; Zhu, X.; et al. The HIF-1α/CXCR4 Pathway Supports Hypoxia-Induced Metastasis of Human Osteosarcoma Cells. Cancer Lett. 2015, 357, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-Inducible Factor Signaling in Pheochromocytoma: Turning the Rudder in the Right Direction. JNCI J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Gao, F.; Yao, Q.; Zhu, J.; Chen, W.; Feng, X.; Feng, B.; Wu, J.; Pacak, K.; Rosenblum, J.; Yu, J.; et al. A Novel HIF2A Mutation Causes Dyslipidemia and Promotes Hepatic Lipid Accumulation. Pharmacol. Res. 2023, 194, 106851. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporters in Cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Ndiaye, P.D.; Dufies, M.; Giuliano, S.; Douguet, L.; Grépin, R.; Durivault, J.; Lenormand, P.; Glisse, N.; Mintcheva, J.; Vouret-Craviari, V.; et al. VEGFC Acts as a Double-Edged Sword in Renal Cell Carcinoma Aggressiveness. Theranostics 2019, 9, 661–675. [Google Scholar] [CrossRef]
- Yang, S.; Liu, L.; Niu, L.; Sun, Y.; Yang, X.; Fan, J.; Ren, J.; Chen, G.G.; Lai, P.B.S. Downregulation and Pro-Apoptotic Effect of Hypoxia-Inducible Factor 2 Alpha in Hepatocellular Carcinoma. Oncotarget 2016, 7, 34571–34581. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.; Wirta, V.; von Stechow, L.; Anne, S.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.-T.; Nistér, M.; et al. A Hypoxic Niche Regulates Glioblastoma Stem Cells through Hypoxia Inducible Factor 2 Alpha. Brain A J. Neurol. 2010, 133, 983–995. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, X.; Liu, W.; Chen, X.; Hou, X.; Shen, D.; Ding, Y.; Yin, J.; Wang, L.; Zhang, H.; et al. Follistatin-like Protein 1 Plays a Tumor Suppressor Role in Clear-Cell Renal Cell Carcinoma. Chin. J. Cancer 2018, 37, 2. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 Regulates Erythropoietic Responses to Hypoxia in Renal Anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial Deletion of Hypoxia-Inducible Factor–2α (HIF-2α) Alters Vascular Function and Tumor Angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Gunaratnam, L.; Morley, M.; Franovic, A.; de Paulsen, N.; Mekhail, K.; Parolin, D.A.E.; Nakamura, E.; Lorimer, I.A.J.; Lee, S. Hypoxia Inducible Factor Activates the Transforming Growth Factor-α/Epidermal Growth Factor Receptor Growth Stimulatory Pathway in VHL-/- Renal Cell Carcinoma Cells*. J. Biol. Chem. 2003, 278, 44966–44974. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Nagamatsu, T.; Morita, K.; Mimura, N.; Iriyama, T.; Fujii, T.; Shibuya, M. HIF-2α, but Not HIF-1α, Mediates Hypoxia-Induced up-Regulation of Flt-1 Gene Expression in Placental Trophoblasts. Sci. Rep. 2018, 8, 17375. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2 Regulates Oct-4: Effects of Hypoxia on Stem Cell Function, Embryonic Development, and Tumor Growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef]
- Jeong, H.-J.; Hong, S.-H.; Park, R.-K.; Shin, T.; An, N.-H.; Kim, H.-M. Hypoxia-Induced IL-6 Production Is Associated with Activation of MAP Kinase, HIF-1, and NF-κB on HEI-OC1 Cells. Hear. Res. 2005, 207, 59–67. [Google Scholar] [CrossRef]
- Wu, J.-W.; Hu, H.; Li, D.; Ma, L.-K. Hypoxia-Inducible Factor 2-Alpha-Dependent Induction of IL-6 Protects the Heart from Ischemia/Reperfusion Injury. Aging 2021, 13, 3443–3458. [Google Scholar] [CrossRef]
- Sena, J.A.; Wang, L.; Pawlus, M.R.; Hu, C.-J. HIFs Enhance the Transcriptional Activation and Splicing of Adrenomedullin. Mol. Cancer Res. 2014, 12, 728–741. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Chen, H.; Mao, Y.; Li, Z.; Kong, C.; Han, B.; Zhang, J.; Chen, Y.; Xue, W.; et al. The Up-regulation of NDRG1 by HIF Counteracts the Cancer-promoting Effect of HIF in VHL-deficient Clear Cell Renal Cell Carcinoma. Cell Prolif. 2020, 53, e12853. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.-J.; Jiang, Y.; Fong, G.-H. Endothelial HIF2α Suppresses Retinal Angiogenesis in Neonatal Mice by Upregulating NOTCH Signaling. Development 2024, 151, dev202802. [Google Scholar] [CrossRef]
- Ortiz-Masià, D.; Cosín-Roger, J.; Calatayud, S.; Hernandez, C.; Alos, R.; Hinojosa, J.; Esplugues, J.; Barrachina, M. M1 Macrophages Activate the Notch Signalling in Epithelial Cells: Relevance in Crohn’s Disease. J. Crohn’s Colitis 2016, 10, 582–592. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Chaudhry, R.; Varacallo, M.A. Biochemistry, Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of Glycolysis by the Hypoxia-inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.A.; Ashcroft, M. Role of Hypoxia-Inducible Factor (HIF)-1A versus HIF-2A in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-Like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- De La Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Gleadle, J.M.; Ratcliffe, P.J. Induction of Hypoxia-Inducible Factor-1, Erythropoietin, Vascular Endothelial Growth Factor, and Glucose Transporter-1 by Hypoxia: Evidence Against a Regulatory Role for Src Kinase. Blood 1997, 89, 503–509. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α- to HIF-2α-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-Inducible Factor 2 Regulates Hepatic Lipid Metabolism. Mol. Cell. Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Hua, X.; Ge, S.; Zhang, L.; Jiang, Q.; Chen, J.; Xiao, H.; Liang, C. MED15 Is Upregulated by HIF-2α and Promotes Proliferation and Metastasis in Clear Cell Renal Cell Carcinoma via Activation of SREBP-Dependent Fatty Acid Synthesis. Cell Death Discov. 2024, 10, 188. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF Drives Lipid Deposition and Cancer in ccRCC via Repression of Fatty Acid Metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1α Induction Suppresses Excessive Lipid Accumulation in Alcoholic Fatty Liver in Mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer. 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative Analysis of Acetyl-CoA Production in Hypoxic Cancer Cells Reveals Substantial Contribution from Acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Li, Y.; Xiao, A.; Lu, Q.; Zeng, H.; Qin, H.; Zheng, E.; Luo, X.; Chen, L.; Ruan, X.Z.; et al. HIF-2α-Induced Upregulation of CD36 Promotes the Development of ccRCC. Exp. Cell Res. 2022, 421, 113389. [Google Scholar] [CrossRef] [PubMed]
- Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The Updated Biology of Hypoxia-Inducible Factor: The Updated Biology of HIF. EMBO J. 2012, 31, 2448–2460. [Google Scholar] [CrossRef]
- Jaśkiewicz, M.; Moszyńska, A.; Króliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzyńska, A.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The Transition from HIF-1 to HIF-2 during Prolonged Hypoxia Results from Reactivation of PHDs and HIF1A mRNA Instability; In Review. Cell. Mol. Biol. Lett 2022, 27, 109. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. HAF: The New Player in Oxygen-Independent HIF-1α Degradation. Cell Cycle 2009, 8, 1359–1366. [Google Scholar] [CrossRef]
- Green, Y.S.; Sargis, T.; Reichert, E.C.; Rudasi, E.; Fuja, D.; Jonasch, E.; Koh, M.Y. Hypoxia-Associated Factor (HAF) Mediates Neurofibromin Ubiquitination and Degradation Leading to Ras–ERK Pathway Activation in Hypoxia. Mol. Cancer Res. 2019, 17, 1220–1232. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 Competes with HSP90 for Binding to HIF-1α and Is Required for O2-Independent and HSP90 Inhibitor-Induced Degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP Selectively Mediate Ubiquitination and Degradation of Hypoxia-Inducible Factor (HIF)-1α but Not HIF-2α. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Serocki, M.; Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan-Jamrozy, K.; Piotrowski, A.; Króliczewski, J.; Collawn, J.F. miR-200b Downregulates Kruppel Like Factor 2 (KLF2) during Acute Hypoxia in Human Endothelial Cells. Eur. J. Cell Biol. 2017, 96, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.P.; Deb, S. (Eds.) Mutant P53 and MDM2 in Cancer. In Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; Volume 85, ISBN 978-94-017-9210-3. [Google Scholar]
- El Azzouzi, H.; Leptidis, S.; Doevendans, P.A.; De Windt, L.J. HypoxamiRs: Regulators of Cardiac Hypoxia and Energy Metabolism. Trends Endocrinol. Metab. 2015, 26, 502–508. [Google Scholar] [CrossRef]
- Curtis, V.F.; Ehrentraut, S.F.; Campbell, E.L.; Glover, L.E.; Bayless, A.; Kelly, C.J.; Kominsky, D.J.; Colgan, S.P. Stabilization of HIF through Inhibition of Cullin-2 Neddylation Is Protective in Mucosal Inflammatory Responses. FASEB J. 2015, 29, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Slawski, J.; Jaśkiewicz, M.; Barton, A.; Kozioł, S.; Collawn, J.F.; Bartoszewski, R. Regulation of the HIF Switch in Human Endothelial and Cancer Cells. Eur. J. Cell Biol. 2024, 103, 151386. [Google Scholar] [CrossRef]
- Jaśkiewicz, M.; Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. EPAS1 Resistance to miRNA-Based Regulation Contributes to Prolonged Expression of HIF-2 during Hypoxia in Human Endothelial Cells. Gene 2023, 868, 147376. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.J.D.; Metcalf, J.L.; Yan, M.S.; Turgeon, P.J.; Wang, J.J.; Chalsev, M.; Petruzziello-Pellegrini, T.N.; Tsui, A.K.Y.; He, J.Z.; Dhamko, H.; et al. Functional Importance of Dicer Protein in the Adaptive Cellular Response to Hypoxia. J. Biol. Chem. 2012, 287, 29003–29020. [Google Scholar] [CrossRef]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF–1α in Response to Hypoxia Is Instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef]
- Pressley, M.; Gallaher, J.A.; Brown, J.S.; Tomaszewski, M.R.; Borad, P.; Damaghi, M.; Gillies, R.J.; Whelan, C.J. Cycling Hypoxia Selects for Constitutive HIF Stabilization. Sci. Rep. 2021, 11, 5777. [Google Scholar] [CrossRef]
- Gillies, R.J.; Brown, J.S.; Anderson, A.R.A.; Gatenby, R.A. Eco-Evolutionary Causes and Consequences of Temporal Changes in Intratumoural Blood Flow. Nat. Rev. Cancer 2018, 18, 576–585. [Google Scholar] [CrossRef]
- Russell, S. Chapter 4—Pseudohypoxia: Life at the Edge. In Ecology and Evolution of Cancer; Academic Press: New York, NY, USA, 2017. [Google Scholar]
- Ramírez-Bergeron, D.L.; Runge, A.; Adelman, D.M.; Gohil, M.; Simon, M.C. HIF-Dependent Hematopoietic Factors Regulate the Development of the Embryonic Vasculature. Dev. Cell 2006, 11, 81–92. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. BioMed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.-H. The Transcription Factor EPAS-1/Hypoxia-Inducible Factor 2α Plays an Important Role in Vascular Remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; VandenBerg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1α Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular Endothelial Growth Factor Induced by Hypoxia May Mediate Hypoxia-Initiated Angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Tarnawski, A.S. Critical Role of Hypoxia Sensor--HIF-1α in VEGF Gene Activation. Implications for Angiogenesis and Tissue Injury Healing. Curr. Med. Chem. 2012, 19, 90–97. [Google Scholar] [CrossRef]
- Shinojima, T.; Oya, M.; Takayanagi, A.; Mizuno, R.; Shimizu, N.; Murai, M. Renal Cancer Cells Lacking Hypoxia Inducible Factor (HIF)-1α Expression Maintain Vascular Endothelial Growth Factor Expression through HIF-2α. Carcinogenesis 2007, 28, 529–536. [Google Scholar] [CrossRef]
- Mailhos, C.; Modlich, U.; Lewis, J.; Harris, A.; Bicknell, R.; Ish-Horowicz, D. Delta4, an Endothelial Specific Notch Ligand Expressed at Sites of Physiological and Tumor Angiogenesis. Differentiation 2001, 69, 135–144. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; Gawrońska-Szklarz, B.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature. Int. J. Mol. Sci. 2021, 22, 843. [Google Scholar] [CrossRef]
- Benwell, C.J.; Johnson, R.T.; Taylor, J.A.G.E.; Price, C.A.; Robinson, S.D. Endothelial VEGFR Coreceptors Neuropilin-1 and Neuropilin-2 Are Essential for Tumor Angiogenesis. Cancer Res. Commun. 2022, 2, 1626–1640. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of Endothelial Cell Metabolism in Vessel Sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef]
- Le Bras, A.; Lionneton, F.; Mattot, V.; Lelièvre, E.; Caetano, B.; Spruyt, N.; Soncin, F. HIF-2alpha Specifically Activates the VE-Cadherin Promoter Independently of Hypoxia and in Synergy with Ets-1 through Two Essential ETS-Binding Sites. Oncogene 2007, 26, 7480–7489. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Kim, G.; Jin, Y.M.; Lee, J.Y.; Shin, J.W.; Jo, I. Expression of Angiopoietin-1 in Hypoxic Pericytes: Regulation by Hypoxia-Inducible Factor-2α and Participation in Endothelial Cell Migration and Tube Formation. Biochem. Biophys. Res. Commun. 2016, 469, 263–269. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin-Tie System. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Yuan, S.; Rigor, R. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. In Regulation of Endothelial Barrier Function; Morgan & Claypool Publishers: San Rafael, CA, USA, 2011; Volume 3, pp. 1–146. [Google Scholar] [CrossRef]
- Yoshida, D.; Kim, K.; Noha, M.; Teramoto, A. Hypoxia Inducible Factor 1-α Regulates of Platelet Derived Growth Factor-B in Human Glioblastoma Cells. J. Neuro-Oncol. 2006, 76, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-Beta in Recruitment of Vascular Smooth Muscle Cells and Pericytes during Embryonic Blood Vessel Formation in the Mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef]
- Li, H.; Huang, H.; Cui, Y.; Li, W.; Zhang, S.; Chen, Y. Study on the Mechanism of Capillary Leakage Caused by Hypoxia-Inducible Factor-1α through Inducing High Expression of Matrix Metalloproteinase-9. J. Oncol. 2021, 2021, 9130650. [Google Scholar] [CrossRef]
- Chan, X.Y.; Volkova, E.; Eoh, J.; Black, R.; Fang, L.; Gorashi, R.; Song, J.; Wang, J.; Elliott, M.B.; Barreto-Ortiz, S.F.; et al. HIF2A Gain-of-Function Mutation Modulates the Stiffness of Smooth Muscle Cells and Compromises Vascular Mechanics. iScience 2021, 24, 102246. [Google Scholar] [CrossRef]
- Ito, K.; Kitajima, Y.; Kai, K.; Matsufuji, S.; Yamada, K.; Egawa, N.; Kitagawa, H.; Okuyama, K.; Tanaka, T.; Noshiro, H. Matrix Metalloproteinase-1 Expression Is Regulated by HIF-1-dependent and Epigenetic Mechanisms and Serves a Tumor-suppressive Role in Gastric Cancer Progression. Int. J. Oncol. 2021, 59, 102. [Google Scholar] [CrossRef]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88. [Google Scholar] [CrossRef]
- Nishi, H.; Sasaki, T.; Nagamitsu, Y.; Terauchi, F.; Nagai, T.; Nagao, T.; Isaka, K. Hypoxia Inducible Factor-1 Mediates Upregulation of Urokinase-Type Plasminogen Activator Receptor Gene Transcription during Hypoxia in Cervical Cancer Cells. Oncol. Rep. 2016, 35, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, J.; Babapoor-Farrokhran, S.; Applewhite, B.; Deshpande, M.; Megarity, H.; Flores-Bellver, M.; Aparicio-Domingo, S.; Ma, T.; Rui, Y.; et al. PAI-1 Is a Vascular Cell-Specific HIF-2-Dependent Angiogenic Factor That Promotes Retinal Neovascularization in Diabetic Patients. Sci. Adv. 2022, 8, eabm1896. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A Receptor Is a Unique Angiogenic Target of HIF-2α in Pulmonary Endothelial Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Lee, S.-S.; Chang, Y.-C. Hypoxic Regulation of Plasminogen Activator Inhibitor-1 Expression in Human Buccal Mucosa Fibroblasts Stimulated with Arecoline. J. Oral. Pathol. Med. 2015, 44, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Jimenez, C.; Armaiz-Pena, G.; Arenillas, C.; Capdevila, J.; Dahia, P.L.M. Hypoxia-Inducible Factor 2 Alpha (HIF2α) Inhibitors: Targeting Genetically Driven Tumor Hypoxia. Endocr. Rev. 2023, 44, 312–322. [Google Scholar] [CrossRef]
- Scheuermann, T.H.; Li, Q.; Ma, H.-W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric Inhibition of Hypoxia Inducible Factor-2 with Small Molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef]
- Fallah, J.; Brave, M.H.; Weinstock, C.; Mehta, G.U.; Bradford, D.; Gittleman, H.; Bloomquist, E.W.; Charlab, R.; Hamed, S.S.; Miller, C.P.; et al. FDA Approval Summary: Belzutifan for von Hippel-Lindau Disease–Associated Tumors. Clin. Cancer Res. 2022, 28, 4843–4848. [Google Scholar] [CrossRef]
- Dutta, D.; Ray, S.; Vivian, J.L.; Paul, S. Activation of the VEGFR1 Chromatin Domain: AN ANGIOGENIC SIGNAL-ETS1/HIF-2α REGULATORY AXIS *. J. Biol. Chem. 2008, 283, 25404–25413. [Google Scholar] [CrossRef]
- Elson, D.A.; Thurston, G.; Huang, L.E.; Ginzinger, D.G.; McDonald, D.M.; Johnson, R.S.; Arbeit, J.M. Induction of Hypervascularity without Leakage or Inflammation in Transgenic Mice Overexpressing Hypoxia-Inducible Factor-1. Genes Dev. 2001, 15, 2520–2532. [Google Scholar] [CrossRef]
- Niemi, H.; Honkonen, K.; Korpisalo, P.; Huusko, J.; Kansanen, E.; Merentie, M.; Rissanen, T.T.; André, H.; Pereira, T.; Poellinger, L.; et al. HIF -1α and HIF -2α Induce Angiogenesis and Improve Muscle Energy Recovery. Eur. J. Clin. Investig. 2014, 44, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-Inducible Factor–Prolyl Hydroxylase Inhibitors in the Treatment of Anemia of Chronic Kidney Disease. Kidney Int. Suppl. 2021, 11, 8–25. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-Inducible Factor–2 (HIF-2) Regulates Hepatic Erythropoietin in Vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hayashi, Y.; Xu, Z.; Wu, L.; Yan, X.; Kalfa, T.A.; Xiao, Z.; Huang, G. Activation of HIF-2a-EPO Axis in Kidney or Liver Is Sufficient to Drive Erythrocytosis in a Novel Inducible HIF-2a Transgenic Mouse Model. Blood 2015, 126, 931. [Google Scholar] [CrossRef]
- Jatho, A.; Zieseniss, A.; Brechtel-Curth, K.; Yamamoto, A.; Coleman, M.L.; Vergel Leon, A.M.; Biggs, D.; Davies, B.; Pugh, C.W.; Ratcliffe, P.J.; et al. Precisely Tuned Inhibition of HIF Prolyl Hydroxylases Is Key for Cardioprotection After Ischemia. Circ. Res. 2021, 128, 1208–1210. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF-1α | HIF-2α | |
|---|---|---|
| Tissue expression | Ubiquitous | Endothelium, hepatocytes, intestinal epithelial cells, pancreatic cells, and alveolar epithelial cells |
| Timing of expression | Acute hypoxia exposure (2–24 h) | Chronic hypoxia exposure (48–72 h) |
| Unique target genes | NRF2 [75], FGF2 [76], ALDH1A [77], CAIX [78], ALDOA [79], ALDOC [80], TPI1 [81], PGK1, ENO A1, PKM2, LDHA, PFK [82], HK1, HK2, GPI, PDK1 [83], iNOS [84], BNIP3 [85], CXCR4 [86], and NRP1 [87] | CCND1 [88], MCT1 [89], VEGFC [90], BAK [91], ABL2 [92], FSTL [93], EPO [94], ANG-2 [95], TGFα [96], FLT1 [97], PLIN2 [88], and OCT4 [98] |
| Common target genes | VEGFA, IL-6 [99,100], ADM [101], NDRG1 [102], CAXII, GLUT1, ADRP [63], MMPs [103], and DLL4 [104] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakleh, M.Z.; Al Haj Zen, A. The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis. Cells 2025, 14, 673. https://doi.org/10.3390/cells14090673
Bakleh MZ, Al Haj Zen A. The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis. Cells. 2025; 14(9):673. https://doi.org/10.3390/cells14090673
Chicago/Turabian StyleBakleh, Mouayad Zuheir, and Ayman Al Haj Zen. 2025. "The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis" Cells 14, no. 9: 673. https://doi.org/10.3390/cells14090673
APA StyleBakleh, M. Z., & Al Haj Zen, A. (2025). The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis. Cells, 14(9), 673. https://doi.org/10.3390/cells14090673