


Inhibitors of Cyclic Dinucleotide Phosphodiesterases and Cyclic Oligonucleotide Ring Nucleases as Potential Drugs for Various Diseases


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
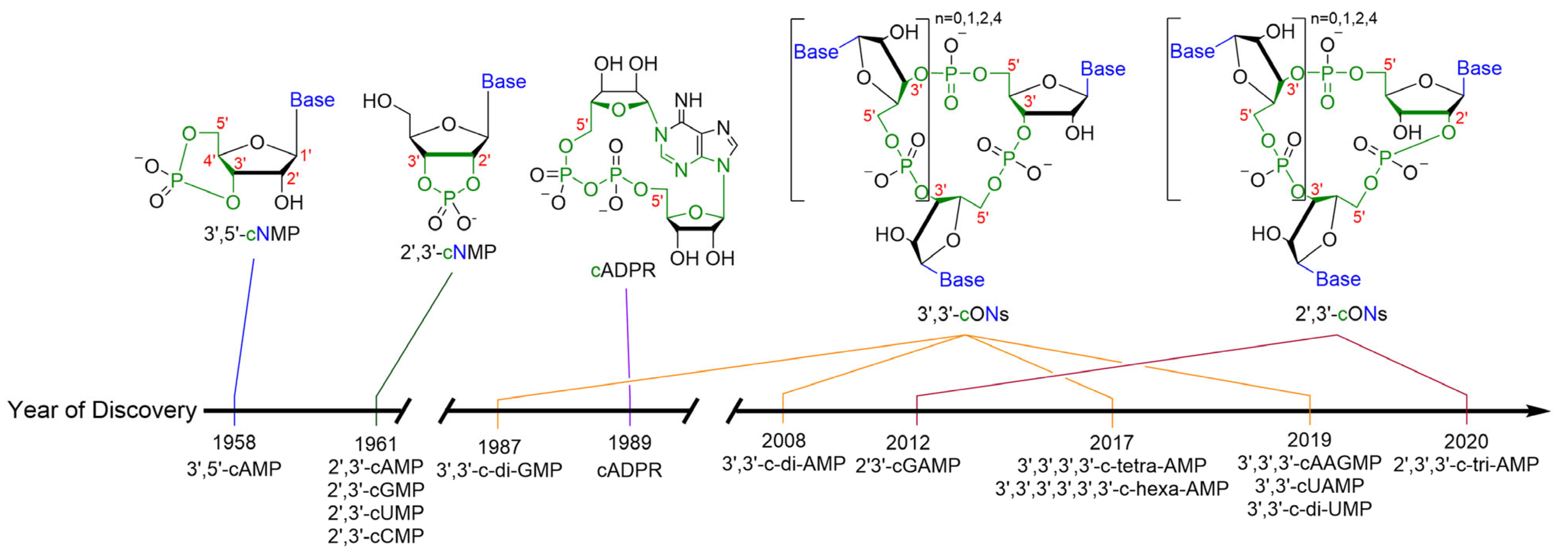
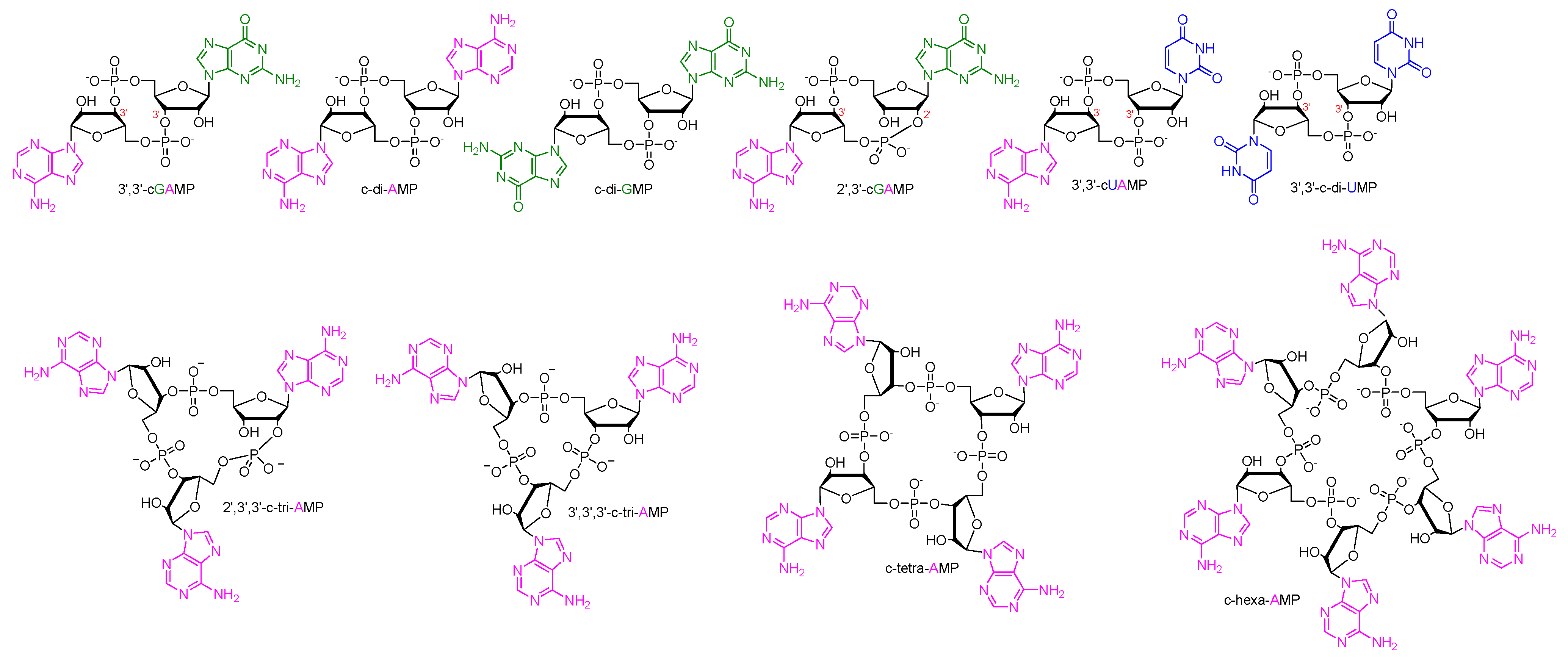
1. Cyclic Oligonucleotides as Signaling Molecules
2. Bacterial cON PDEs as Potential Drug Targets
2.1. Targeting Bacterial Cyclic-di-GMP PDEs
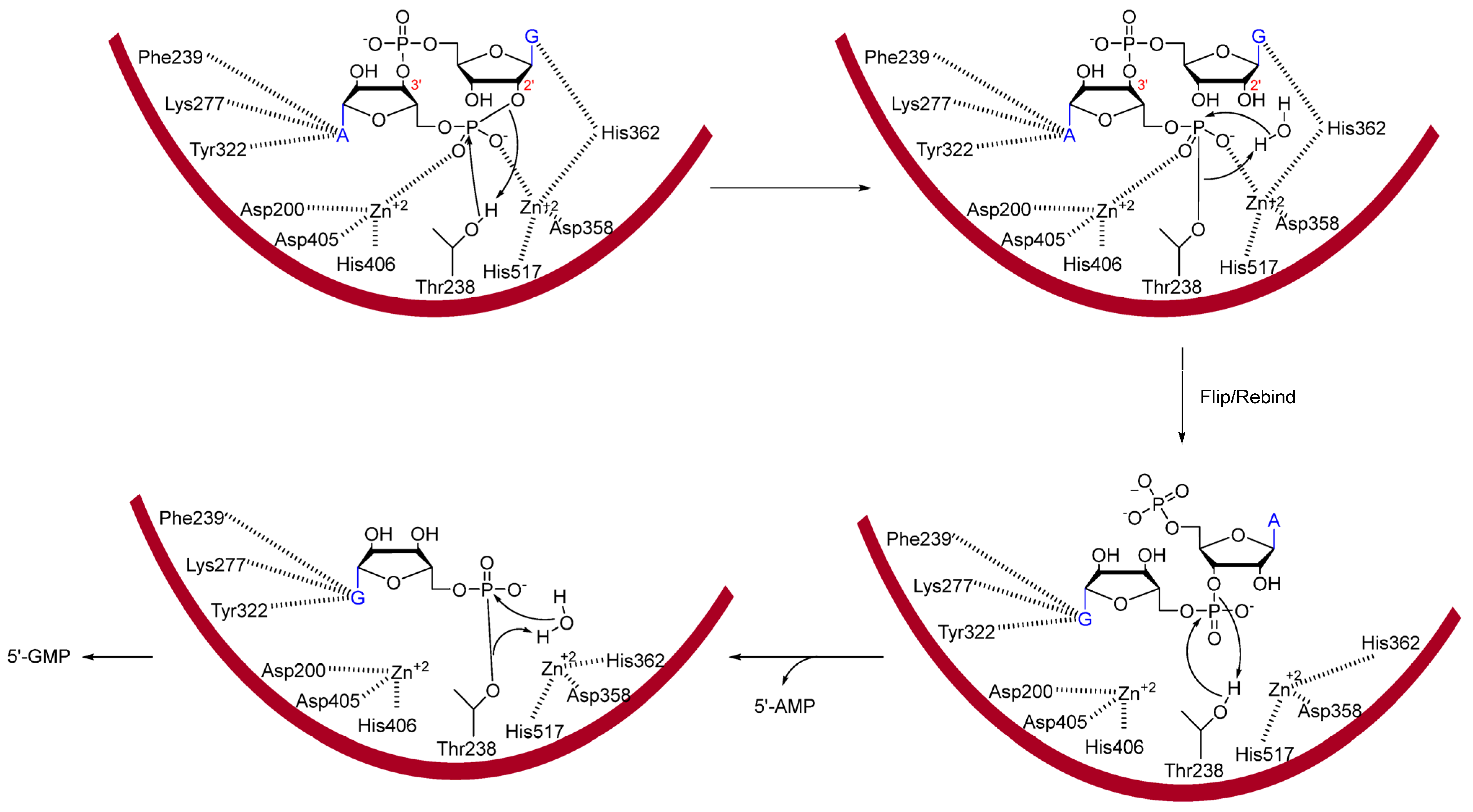
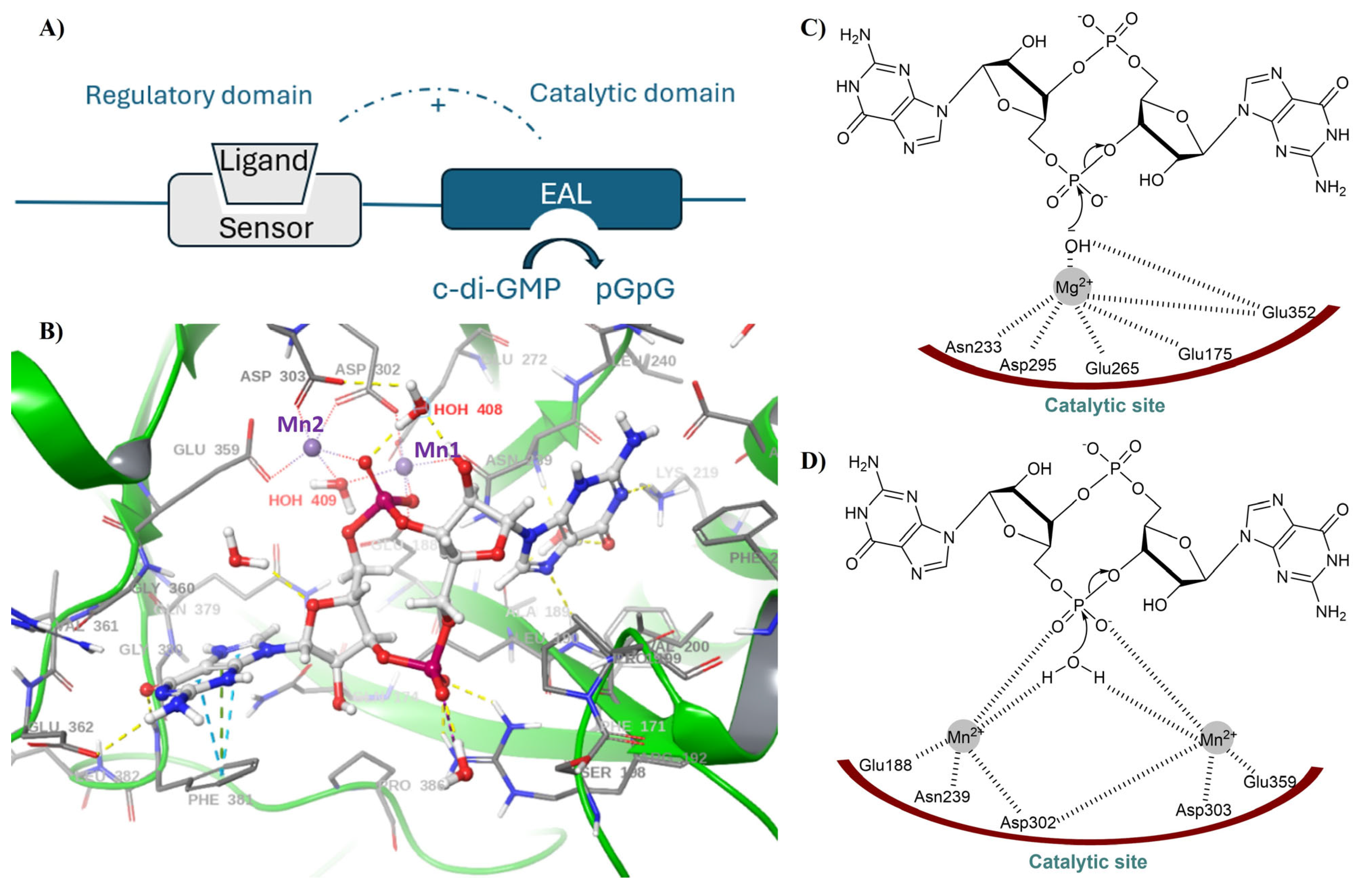
2.2. EAL PDEs

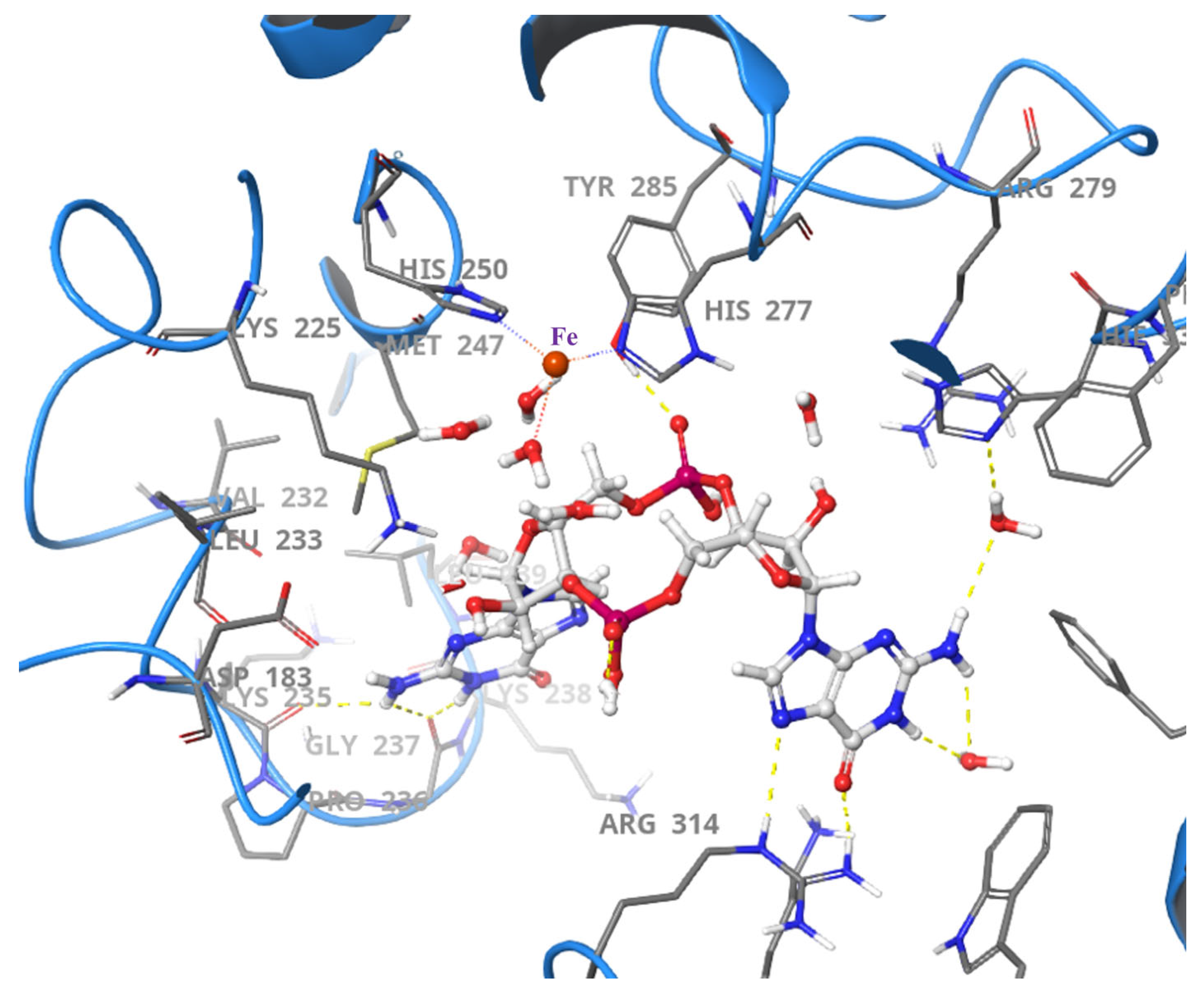
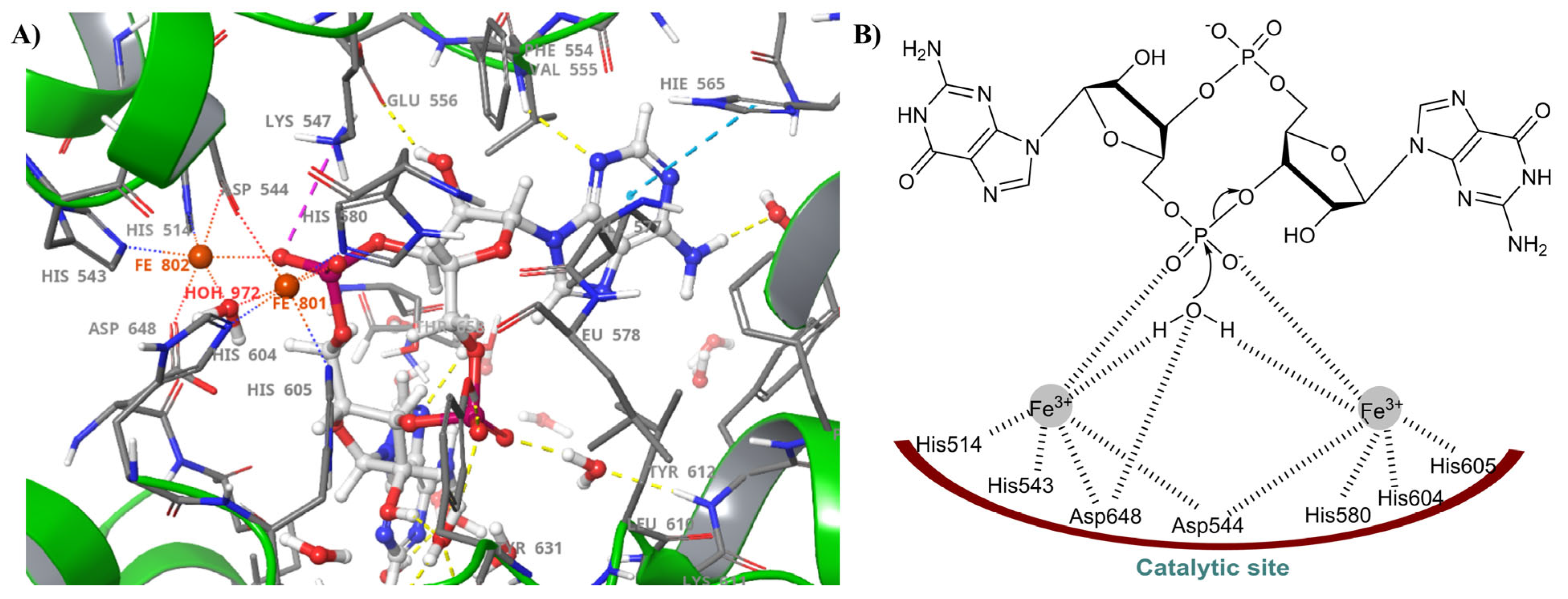
2.3. HD-GYP PDEs
3. Development of Cyclic-di-GMP PDEs Inhibitors
4. Therapeutic Agents Against P. aeruginosa Infections
5. Attenuation of Adherent-Invasive Escherichia coli Pathogenicity in Crohn’s Disease
6. Potential Anti-Virulence Agents for the Treatment of Cholera Disease
7. Potential Anti-Virulence Agents for Black Rot Disease in Plants
8. Targeting c-di-AMP PDEs
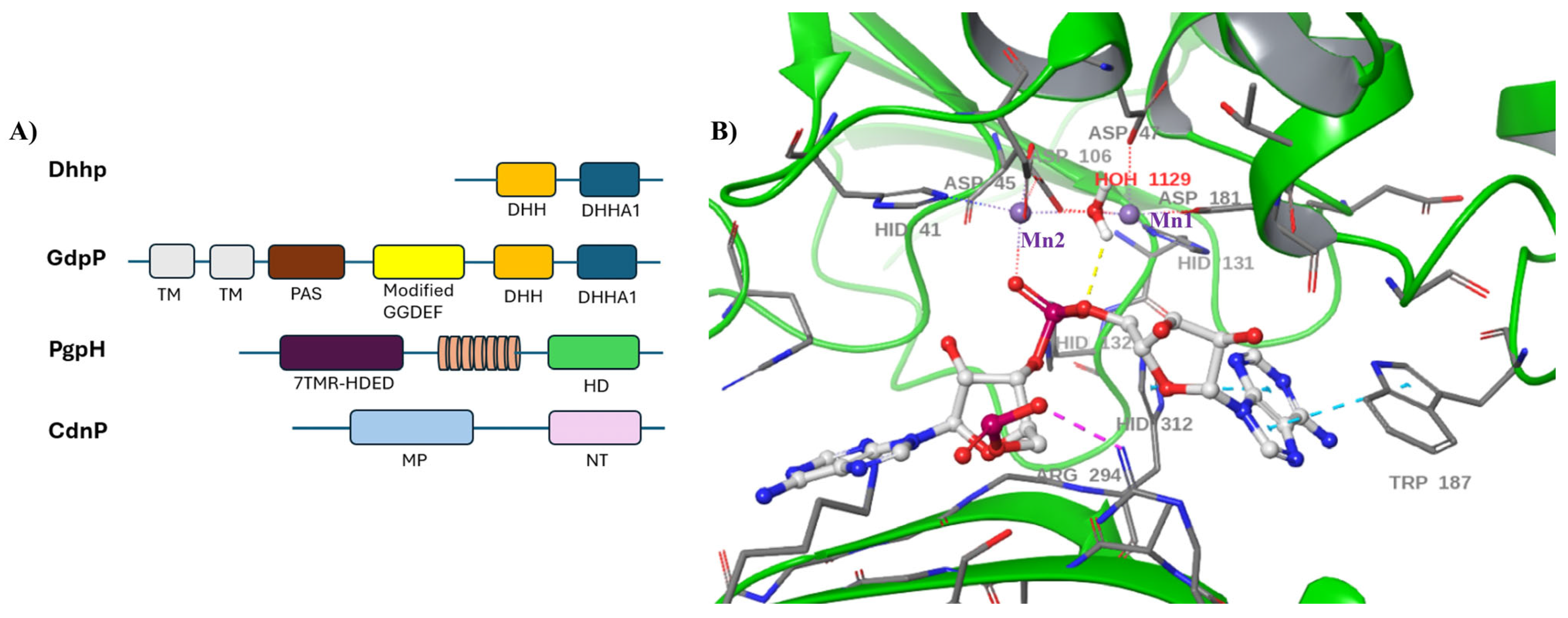
9. Dhhp-Type Phosphodiesterase

10. CdnP-Type Phosphodiesterases
11. PgpH and GdpP-Type Phosphodiesterases
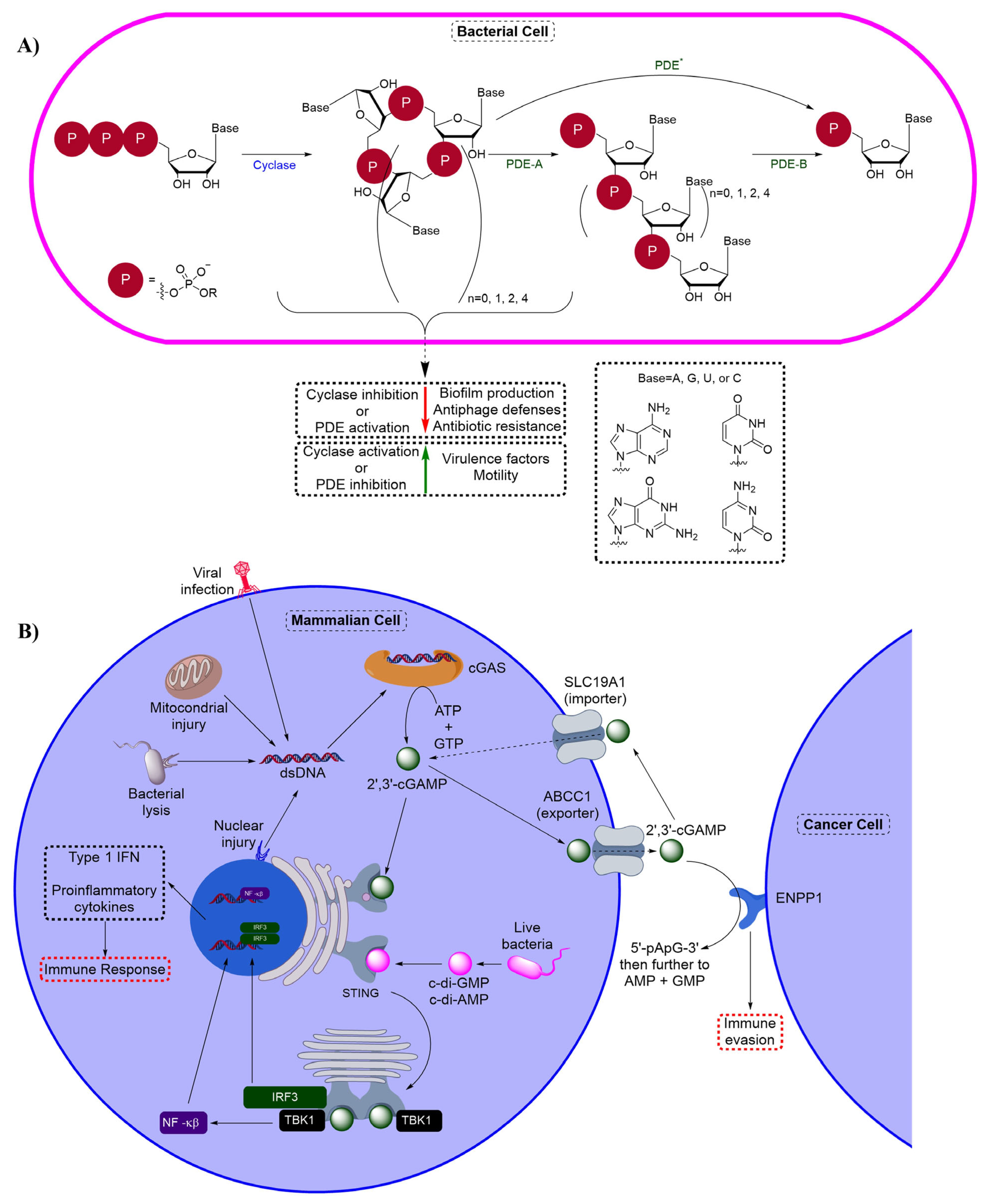
12. Bacterial Anti-Phage Defenses
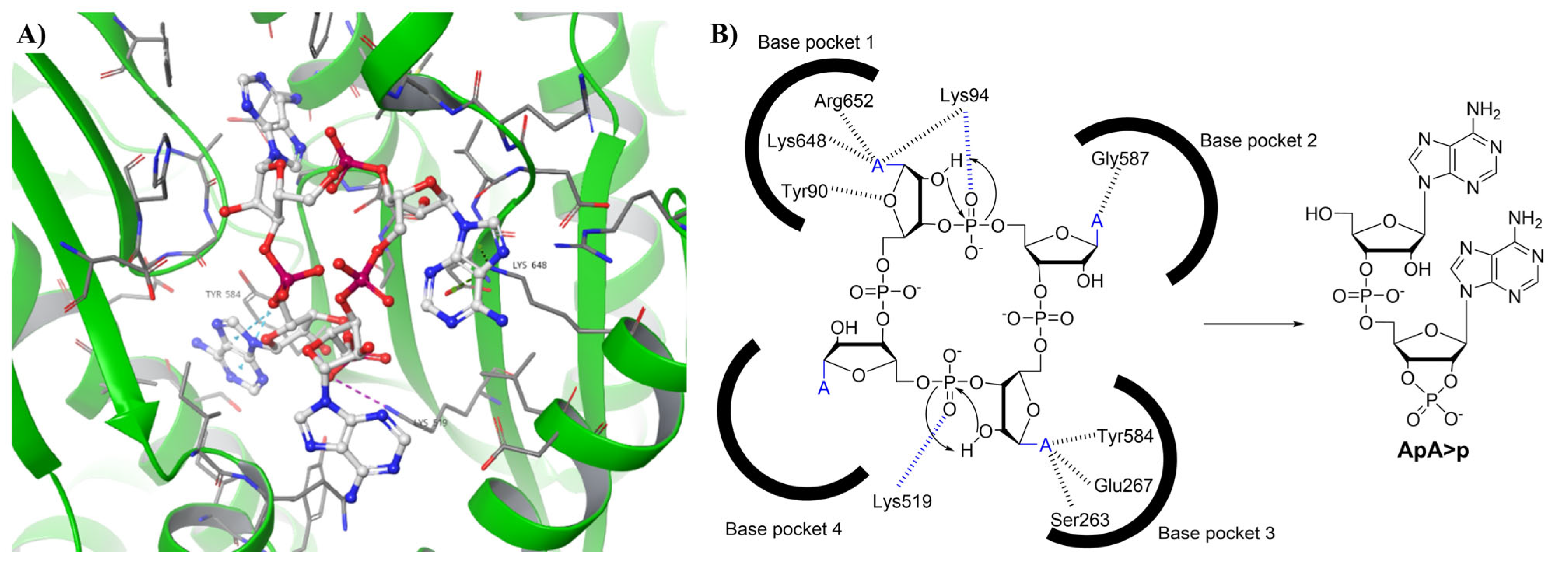
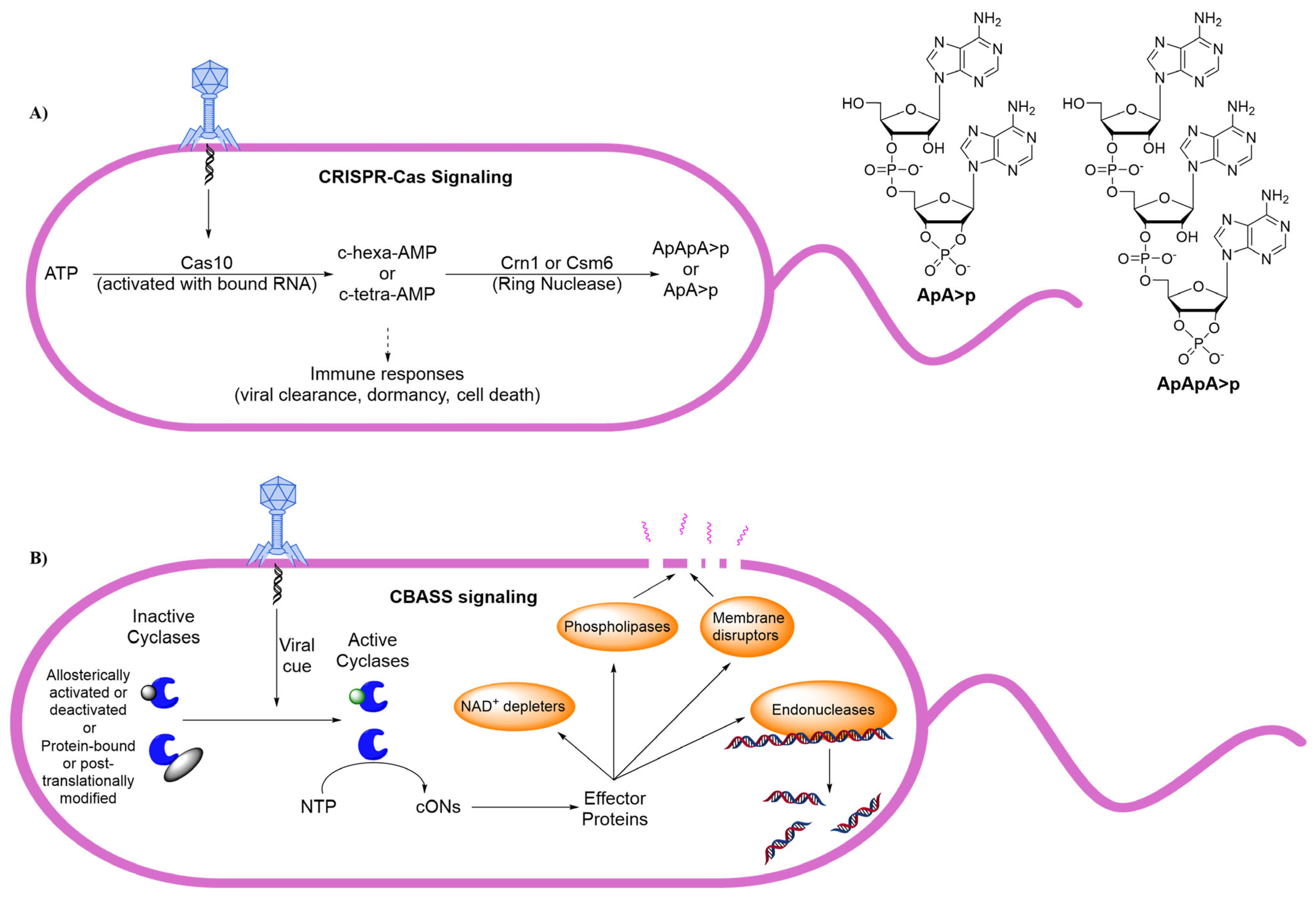
13. Human cON PDEs as Potential Drug Targets
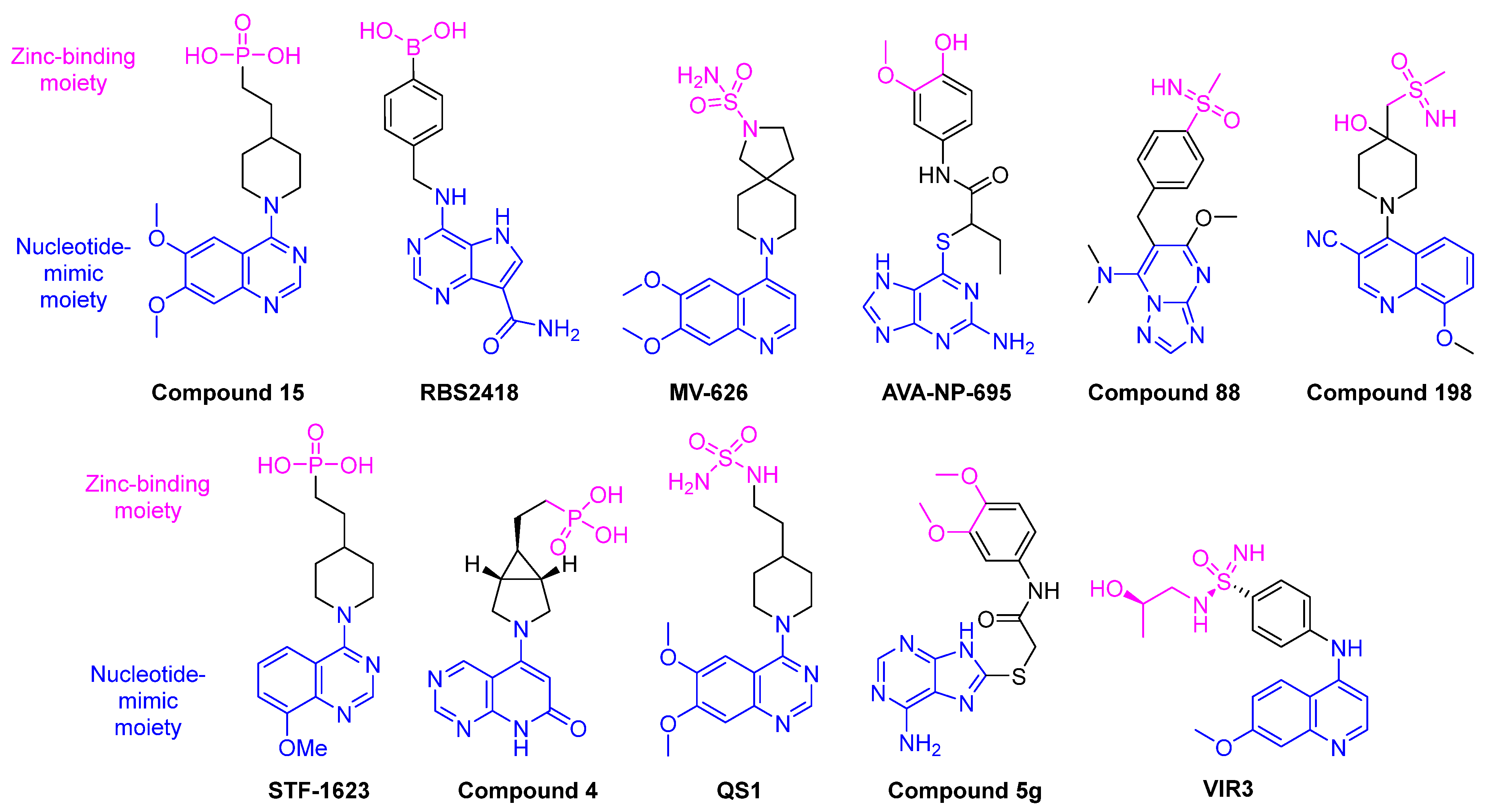
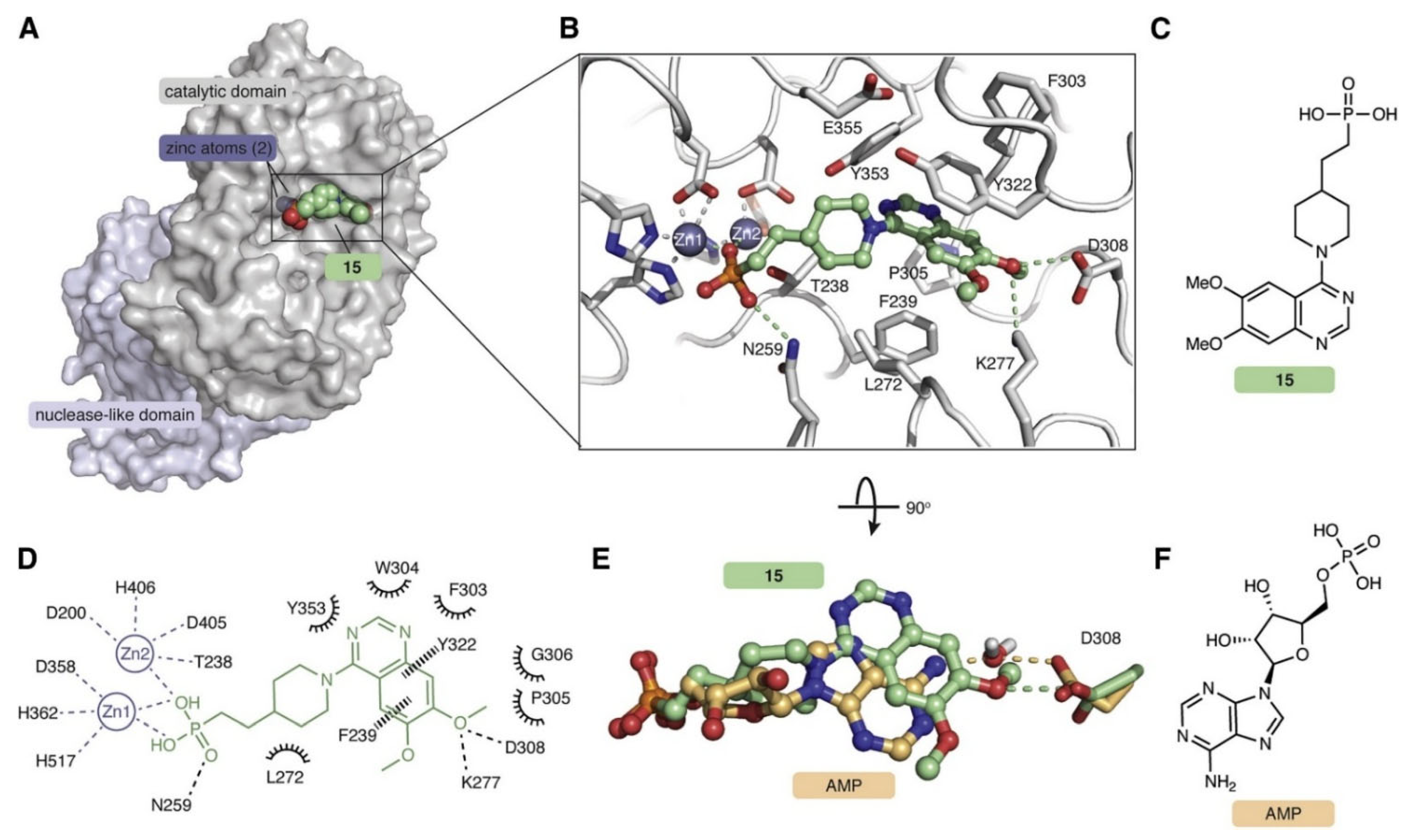
14. Disappearance of cONs in Fungi
15. Future Prospects
16. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.A.; Lie, J.D. Phosphodiesterase-5 (PDE5) Inhibitors in the Management of Erectile Dysfunction. Pharm. Ther. 2013, 38, 407–419. [Google Scholar]
- Zuo, H.; Cattani-Cavalieri, I.; Musheshe, N.; Nikolaev, V.O.; Schmidt, M. Phosphodiesterases as Therapeutic Targets for Respiratory Diseases. Pharmacol. Ther. 2019, 197, 225–242. [Google Scholar] [CrossRef]
- Leroy, J.; Fischmeister, R. Inhibit a Phosphodiesterase to Treat Heart Failure? Circulation 2018, 138, 2003–2006. [Google Scholar] [CrossRef]
- Tzoumas, N.; Farrah, T.E.; Dhaun, N.; Webb, D.J. Established and Emerging Therapeutic Uses of PDE Type 5 Inhibitors in Cardiovascular Disease. Br. J. Pharmacol. 2020, 177, 5467–5488. [Google Scholar] [CrossRef]
- Lusardi, M.; Rapetti, F.; Spallarossa, A.; Brullo, C. PDE4D: A Multipurpose Pharmacological Target. Int. J. Mol. Sci. 2024, 25, 8052. [Google Scholar] [CrossRef]
- Kamel, R.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. Cyclic Nucleotide Phosphodiesterases as Therapeutic Targets in Cardiac Hypertrophy and Heart Failure. Nat. Rev. Cardiol. 2023, 20, 90–108. [Google Scholar] [CrossRef]
- Nemr, M.T.M.; Abdelaziz, M.A.; Teleb, M.; Elmasry, A.E.; Elshaier, Y.A.A.M. An Overview on Pharmaceutical Applications of Phosphodiesterase Enzyme 5 (PDE5) Inhibitors. In Molecular Diversity; Springer Nature: London, UK, 2024. [Google Scholar] [CrossRef]
- Ross, P.; Weinhouse, H.; Aloni, Y.; Michaeli, D.; Weinberger-Ohana, P.; Mayer, R.; Braun, S.; de Vroom, E.; van der Marel, G.A.; van Boom, J.H.; et al. Regulation of Cellulose Synthesis in Acetobacter Xylinum by Cyclic Diguanylic Acid. Nature 1987, 325, 279–281. [Google Scholar] [CrossRef]
- Ross, P.; Aloni, Y.; Weinhouse, C.; Michaeli, D.; Weinberger-Ohana, P.; Meyer, R.; Benziman, M. An Unusual Guanyl Oligonucleotide Regulates Cellulose Synthesis in Acetobacter Xylinum. FEBS Lett. 1985, 186, 191–196. [Google Scholar] [CrossRef]
- Gomelsky, M. cAMP, c-di-GMP, c-di-AMP and Now cGMP: Bacteria Use Them All! Mol. Microbiol. 2011, 79, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Danilchanka, O.; Mekalanos, J.J. Cyclic Dinucleotides and the Innate Immune Response. Cell 2013, 154, 962–970. [Google Scholar] [CrossRef]
- Wade, H.E. The Autodegradation of Ribonucleoprotein in Escherichia Coli. Biochem. J. 1961, 78, 457–472. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural Determination of a Cyclic Metabolite of NAD+ with Intracellular Ca2+-Mobilizing Activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef]
- Witte, G.; Hartung, S.; Büttner, K.; Hopfner, K.-P. Structural Biochemistry of a Bacterial Checkpoint Protein Reveals Diadenylate Cyclase Activity Regulated by DNA Recombination Intermediates. Mol. Cell 2008, 30, 167–178. [Google Scholar] [CrossRef]
- Kazlauskiene, M.; Kostiuk, G.; Venclovas, Č.; Tamulaitis, G.; Siksnys, V. A Cyclic Oligonucleotide Signaling Pathway in Type III CRISPR-Cas Systems. Science 2017, 357, 605–609. [Google Scholar] [CrossRef]
- Niewoehner, O.; Garcia-Doval, C.; Rostøl, J.T.; Berk, C.; Schwede, F.; Bigler, L.; Hall, J.; Marraffini, L.A.; Jinek, M. Type III CRISPR–Cas Systems Produce Cyclic Oligoadenylate Second Messengers. Nature 2017, 548, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, A.T.; Eaglesham, J.B.; de Oliveira Mann, C.C.; Morehouse, B.R.; Lowey, B.; Nieminen, E.A.; Danilchanka, O.; King, D.S.; Lee, A.S.Y.; Mekalanos, J.J.; et al. Bacterial cGAS-like Enzymes Synthesize Diverse Nucleotide Signals. Nature 2019, 567, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Lowey, B.; Whiteley, A.T.; Keszei, A.F.A.; Morehouse, B.R.; Mathews, I.T.; Antine, S.P.; Cabrera, V.J.; Kashin, D.; Niemann, P.; Jain, M.; et al. CBASS Immunity Uses CARF-Related Effectors to Sense 3′–5′- and 2′–5′-Linked Cyclic Oligonucleotide Signals and Protect Bacteria from Phage Infection. Cell 2020, 182, 38–49.e17. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent Functional Insights into the Role of (p)ppGpp in Bacterial Physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef]
- Ying, W.; Zhao, D.; Ouyang, P.; Subramanya, V.; Vaidya, D.; Ndumele, C.E.; Guallar, E.; Sharma, K.; Shah, S.J.; Kass, D.A.; et al. Associations Between the Cyclic Guanosine Monophosphate Pathway and Cardiovascular Risk Factors: MESA. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2019, 8, e013149. [Google Scholar] [CrossRef]
- McDonough, K.A.; Rodriguez, A. The Myriad Roles of Cyclic AMP in Microbial Pathogens: From Signal to Sword. Nat. Rev. Microbiol. 2012, 10, 27–38. [Google Scholar] [CrossRef]
- Chauhan, S.S.; Marotta, N.J.; Karls, A.C.; Weinert, E.E. Binding of 2′,3′-Cyclic Nucleotide Monophosphates to Bacterial Ribosomes Inhibits Translation. ACS Cent. Sci. 2022, 8, 1518–1526. [Google Scholar] [CrossRef]
- Marotta, N.J.; Weinert, E.E. Insights into the Metabolism, Signaling, and Physiological Effects of 2′,3′-Cyclic Nucleotide Monophosphates in Bacteria. Crit. Rev. Biochem. Mol. Biol. 2023, 58, 118–131. [Google Scholar] [CrossRef]
- Ahn, J.; Barber, G.N. STING Signaling and Host Defense against Microbial Infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krasteva, P.V.; Sondermann, H. Versatile Modes of Cellular Regulation via Cyclic Dinucleotides. Nat. Chem. Biol. 2017, 13, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Hengge, R. Principles of C-di-GMP Signalling in Bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273. [Google Scholar] [CrossRef]
- Wolfe, A.J.; Visick, K.L. Get the Message Out: Cyclic-di-GMP Regulates Multiple Levels of Flagellum-Based Motility. J. Bacteriol. 2008, 190, 463–475. [Google Scholar] [CrossRef]
- Opoku-Temeng, C.; Zhou, J.; Zheng, Y.; Su, J.; Sintim, H.O. Cyclic Dinucleotide (c-di-GMP, c-di-AMP, and cGAMP) Signalings Have Come of Age to Be Inhibited by Small Molecules. Chem. Commun. 2016, 52, 9327–9342. [Google Scholar] [CrossRef]
- Orr, M.W.; Donaldson, G.P.; Severin, G.B.; Wang, J.; Sintim, H.O.; Waters, C.M.; Lee, V.T. Oligoribonuclease is the Primary Degradative Enzyme for pGpG in Pseudomonas aeruginosa That Is Required for Cyclic-di-GMP Turnover. Proc. Natl. Acad. Sci. USA 2015, 112, E5048–E5057. [Google Scholar] [CrossRef]
- Christen, M.; Christen, B.; Folcher, M.; Schauerte, A.; Jenal, U. Identification and Characterization of a Cyclic di-GMP-Specific Phosphodiesterase and Its Allosteric Control by GTP. J. Biol. Chem. 2005, 280, 30829–30837. [Google Scholar] [CrossRef]
- Chen, H.-J.; Li, N.; Luo, Y.; Jiang, Y.-L.; Zhou, C.-Z.; Chen, Y.; Li, Q. The GDP-Switched GAF Domain of DcpA Modulates the Concerted Synthesis/Hydrolysis of c-di-GMP in Mycobacterium Smegmatis. Biochem. J. 2018, 475, 1295–1308. [Google Scholar] [CrossRef]
- Galperin, M.Y. Structural Classification of Bacterial Response Regulators: Diversity of Output Domains and Domain Combinations. J. Bacteriol. 2006, 188, 4169–4182. [Google Scholar] [CrossRef]
- Liu, C.; Liew, C.W.; Wong, Y.H.; Tan, S.T.; Poh, W.H.; Manimekalai, M.S.S.; Rajan, S.; Xin, L.; Liang, Z.-X.; Grüber, G.; et al. Insights into Biofilm Dispersal Regulation from the Crystal Structure of the PAS-GGDEF-EAL Region of RbdA from Pseudomonas aeruginosa. J. Bacteriol. 2018, 200, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Beyhan, S.; Yildiz, F.H. Regulation of Vibrio Polysaccharide Synthesis and Virulence Factor Production by CdgC, a GGDEF-EAL Domain Protein, in Vibrio Cholerae. J. Bacteriol. 2007, 189, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.V.A.S.; De, N.; Bae, N.; Wang, Q.; Sondermann, H. Structural Analysis of the GGDEF-EAL Domain-Containing c-di-GMP Receptor FimX. Structure 2009, 17, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; Hartmann, E.; Griese, J.J.; Beitlich, T.; Kirienko, N.V.; Ryjenkov, D.A.; Reinstein, J.; Shoeman, R.L.; Gomelsky, M.; Schlichting, I. Structure and Mechanism of a Bacterial Light-Regulated Cyclic Nucleotide Phosphodiesterase. Nature 2009, 459, 1015–1018. [Google Scholar] [CrossRef]
- Rao, F.; Yang, Y.; Qi, Y.; Liang, Z.-X. Catalytic Mechanism of Cyclic di-GMP-Specific Phosphodiesterase: A Study of the EAL Domain-Containing RocR from Pseudomonas aeruginosa. J. Bacteriol. 2008, 190, 3622–3631. [Google Scholar] [CrossRef]
- Salter, E.A.; Wierzbicki, A. The Mechanism of Cyclic Nucleotide Hydrolysis in the Phosphodiesterase Catalytic Site. J. Phys. Chem. B 2007, 111, 4547–4552. [Google Scholar] [CrossRef]
- Xiong, Y.; Lu, H.-T.; Li, Y.; Yang, G.-F.; Zhan, C.-G. Characterization of a Catalytic Ligand Bridging Metal Ions in Phosphodiesterases 4 and 5 by Molecular Dynamics Simulations and Hybrid Quantum Mechanical/Molecular Mechanical Calculations. Biophys. J. 2006, 91, 1858–1867. [Google Scholar] [CrossRef]
- Tchigvintsev, A.; Xu, X.; Singer, A.; Chang, C.; Brown, G.; Proudfoot, M.; Cui, H.; Flick, R.; Anderson, W.F.; Joachimiak, A.; et al. Structural Insight into the Mechanism of C-di-GMP Hydrolysis by EAL Domain Phosphodiesterases. J. Mol. Biol. 2010, 402, 524–538. [Google Scholar] [CrossRef]
- Bellini, D.; Horrell, S.; Hutchin, A.; Phippen, C.W.; Strange, R.W.; Cai, Y.; Wagner, A.; Webb, J.S.; Tews, I.; Walsh, M.A. Dimerisation Induced Formation of the Active Site and the Identification of Three Metal Sites in EAL-Phosphodiesterases. Sci. Rep. 2017, 7, 42166. [Google Scholar] [CrossRef]
- Sundriyal, A.; Massa, C.; Samoray, D.; Zehender, F.; Sharpe, T.; Jenal, U.; Schirmer, T. Inherent Regulation of EAL Domain-Catalyzed Hydrolysis of Second Messenger Cyclic di-GMP. J. Biol. Chem. 2014, 289, 6978–6990. [Google Scholar] [CrossRef]
- Bellini, D.; Hutchin, A.; Soren, O.; Webb, J.S.; Tews, I.; Walsh, M.A. Structure and Regulation of EAL Domain Proteins. In Microbial Cyclic di-Nucleotide Signaling; Chou, S.-H., Guiliani, N., Lee, V.T., Römling, U., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 27–48. [Google Scholar] [CrossRef]
- Filippova, E.V.; Minasov, G.; Shuvalova, L.; Kiryukhina, O.; Massa, C.; Schirmer, T.; Joachimiak, A.; Anderson, W.F. RCSB PDB—3U2E: EAL Domain of Phosphodiesterase PdeA in Complex with 5′-pGpG and Mg++. Available online: https://www.rcsb.org/structure/3u2e (accessed on 17 March 2025).
- Galperin, M.Y.; Natale, D.A.; Aravind, L.; Koonin, E.V. A Specialized Version of the HD Hydrolase Domain Implicated in Signal Transduction. J. Mol. Microbiol. Biotechnol. 1999, 1, 303–305. [Google Scholar] [PubMed]
- Galperin, M.Y. A Census of Membrane-Bound and Intracellular Signal Transduction Proteins in Bacteria: Bacterial IQ, Extroverts and Introverts. BMC Microbiol. 2005, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, C.; Jiang, W.; Wang, L.; Li, C.; Wang, Y.; Dow, J.M.; Sun, W. The HD-GYP Domain Protein RpfG of Xanthomonas Oryzae Pv. Oryzicola Regulates Synthesis of Extracellular Polysaccharides That Contribute to Biofilm Formation and Virulence on Rice. PLoS ONE 2013, 8, e59428. [Google Scholar] [CrossRef] [PubMed]
- McKee, R.W.; Kariisa, A.; Mudrak, B.; Whitaker, C.; Tamayo, R. A Systematic Analysis of the in Vitro and in Vivo Functions of the HD-GYP Domain Proteins of Vibrio Cholerae. BMC Microbiol. 2014, 14, 272. [Google Scholar] [CrossRef]
- Ryan, R.P.; Lucey, J.; O’Donovan, K.; McCarthy, Y.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. HD-GYP Domain Proteins Regulate Biofilm Formation and Virulence in Pseudomonas aeruginosa. Environ. Microbiol. 2009, 11, 1126–1136. [Google Scholar] [CrossRef]
- Sultan, S.Z.; Pitzer, J.E.; Boquoi, T.; Hobbs, G.; Miller, M.R.; Motaleb, M.A. Analysis of the HD-GYP Domain Cyclic Dimeric GMP Phosphodiesterase Reveals a Role in Motility and the Enzootic Life Cycle of Borrelia Burgdorferi. Infect. Immun. 2011, 79, 3273–3283. [Google Scholar] [CrossRef]
- Deng, M.; Tao, J.; Ye, Z.Y.; Jiang, Z.; Yu, J.; Su, X. Novel Mechanism for Cyclic Dinucleotide Degradation Revealed by Structural Studies of Vibrio Phosphodiesterase V-cGAP3. J. Mol. Biol. 2018, 430, 5080–5093. [Google Scholar] [CrossRef]
- Miner, K.D.; Klose, K.E.; Kurtz, D.M., Jr. An HD-GYP Cyclic di-Guanosine Monophosphate Phosphodiesterase with a Non-Heme Diiron–Carboxylate Active Site. Biochemistry 2013, 52, 5329–5331. [Google Scholar] [CrossRef]
- Bellini, D.; Caly, D.L.; McCarthy, Y.; Bumann, M.; An, S.-Q.; Dow, J.M.; Ryan, R.P.; Walsh, M.A. Crystal Structure of an HD-GYP Domain Cyclic-di-GMP Phosphodiesterase Reveals an Enzyme with a Novel Trinuclear Catalytic Iron Centre. Mol. Microbiol. 2014, 91, 26–38. [Google Scholar] [CrossRef]
- Rinaldo, S.; Paiardini, A.; Paone, A.; Cutruzzolà, F.; Giardina, G. Structure and Function of HD-GYP Phosphodiesterases. In Microbial Cyclic di-Nucleotide Signaling; Chou, S.-H., Guiliani, N., Lee, V.T., Römling, U., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 65–78. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef]
- Zheng, Y.; Tsuji, G.; Opoku-Temeng, C.; Sintim, H.O. Inhibition of P. Aeruginosa c-di-GMP Phosphodiesterase RocR and Swarming Motility by a Benzoisothiazolinone Derivative. Chem. Sci. 2016, 7, 6238–6244. [Google Scholar] [CrossRef] [PubMed]
- Kulesekara, H.; Lee, V.; Brencic, A.; Liberati, N.; Urbach, J.; Miyata, S.; Lee, D.G.; Neely, A.N.; Hyodo, M.; Hayakawa, Y.; et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. USA 2006, 103, 2839–2844. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- da Silva Filho, L.V.R.F.; de Ferreira, F.A.; Reis, F.J.C.; de Britto, M.C.A.; Levy, C.E.; Clark, O.; Ribeiro, J.D. Pseudomonas aeruginosa Infection in Patients with Cystic Fibrosis: Scientific Evidence Regarding Clinical Impact, Diagnosis, and Treatment. J. Bras. Pneumol. 2013, 39, 495–512. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Garcia-Gil, L.J. Escherichia coli in Chronic Inflammatory Bowel Diseases: An Update on Adherent Invasive Escherichia coli Pathogenicity. World J. Gastrointest. Pathophysiol. 2014, 5, 213–227. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial Influences in Inflammatory Bowel Diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Claret, L.; Miquel, S.; Vieille, N.; Ryjenkov, D.A.; Gomelsky, M.; Darfeuille-Michaud, A. The Flagellar Sigma Factor FliA Regulates Adhesion and Invasion of Crohn Disease-Associated Escherichia coli via a Cyclic Dimeric GMP-Dependent Pathway. J. Biol. Chem. 2007, 282, 33275–33283. [Google Scholar] [CrossRef]
- Bomchil, N.; Watnick, P.; Kolter, R. Identification and Characterization of a Vibrio Cholerae Gene, mbaA, Involved in Maintenance of Biofilm Architecture. J. Bacteriol. 2003, 185, 1384–1390. [Google Scholar] [CrossRef]
- Yildiz, F.H.; Liu, X.S.; Heydorn, A.; Schoolnik, G.K. Molecular Analysis of Rugosity in a Vibrio Cholerae O1 El Tor Phase Variant. Mol. Microbiol. 2004, 53, 497–515. [Google Scholar] [CrossRef]
- Karatan, E.; Duncan, T.R.; Watnick, P.I. NspS, a Predicted Polyamine Sensor, Mediates Activation of Vibrio cholerae Biofilm Formation by Norspermidine. J. Bacteriol. 2005, 187, 7434–7443. [Google Scholar] [CrossRef]
- Tischler, A.D.; Camilli, A. Cyclic Diguanylate (c-di-GMP) Regulates Vibrio Cholerae Biofilm Formation. Mol. Microbiol. 2004, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.E.; Tang, J.L.; Feng, J.X.; Pan, M.Q.; Wilson, T.J.G.; Slater, H.; Dow, J.M.; Williams, P.; Daniels, M.J. A Novel Regulatory System Required for Pathogenicity of Xanthomonas campestris Is Mediated by a Small Diffusible Signal Molecule. Mol. Microbiol. 1997, 24, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.P.; Fouhy, Y.; Lucey, J.F.; Jiang, B.-L.; He, Y.-Q.; Feng, J.-X.; Tang, J.-L.; Dow, J.M. Cyclic di-GMP Signalling in the Virulence and Environmental Adaptation of Xanthomonas campestris. Mol. Microbiol. 2007, 63, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Jia, Y.; Ren, S.-X.; He, Y.-Q.; Feng, J.-X.; Lu, L.-F.; Sun, Q.; Ying, G.; Tang, D.-J.; Tang, H.; et al. Comparative and Functional Genomic Analyses of the Pathogenicity of Phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 2005, 15, 757–767. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, Y.; Bai, G.; Zhou, X.; Wu, H. Cyclic di-AMP Mediates Biofilm Formation. Mol. Microbiol. 2016, 99, 945–959. [Google Scholar] [CrossRef]
- Commichau, F.M.; Heidemann, J.L.; Ficner, R.; Stülke, J. Making and Breaking of an Essential Poison: The Cyclases and Phosphodiesterases That Produce and Degrade the Essential Second Messenger Cyclic di-AMP in Bacteria. J. Bacteriol. 2019, 201, 10-1128. [Google Scholar] [CrossRef]
- Gundlach, J.; Mehne, F.M.P.; Herzberg, C.; Kampf, J.; Valerius, O.; Kaever, V.; Stülke, J. An Essential Poison: Synthesis and Degradation of Cyclic di-AMP in Bacillus Subtilis. J. Bacteriol. 2015, 197, 3265–3274. [Google Scholar] [CrossRef]
- Rao, F.; See, R.Y.; Zhang, D.; Toh, D.C.; Ji, Q.; Liang, Z.-X. YybT Is a Signaling Protein That Contains a Cyclic Dinucleotide Phosphodiesterase Domain and a GGDEF Domain with ATPase Activity. J. Biol. Chem. 2010, 285, 473–482. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Abbott, J.C.; Burhenne, H.; Kaever, V.; Gründling, A. C-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress. PLoS Pathog. 2011, 7, e1002217. [Google Scholar] [CrossRef]
- Liu, S.; Bayles, D.O.; Mason, T.M.; Wilkinson, B.J. A Cold-Sensitive Listeria Monocytogenes Mutant Has a Transposon Insertion in a Gene Encoding a Putative Membrane Protein and Shows Altered (p)ppGpp Levels. Appl. Environ. Microbiol. 2006, 72, 3955–3959. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, J.; Eisele, L.E.; Underwood, A.J.; Koestler, B.J.; Waters, C.M.; Metzger, D.W.; Bai, G. Two DHH Subfamily 1 Proteins in Streptococcus pneumoniae Possess Cyclic di-AMP Phosphodiesterase Activity and Affect Bacterial Growth and Virulence. J. Bacteriol. 2013, 195, 5123–5132. [Google Scholar] [CrossRef] [PubMed]
- Andrade, W.A.; Firon, A.; Schmidt, T.; Hornung, V.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Trieu-Cuot, P.; Golenbock, D.T.; Kaminski, P.-A. Group B Streptococcus Degrades Cyclic-di-AMP to Modulate STING-Dependent Type I Interferon Production. Cell Host Microbe 2016, 20, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Zhang, J.-J.; Fang, X.; Lawlis, G.B.; Troxell, B.; Zhou, Y.; Gomelsky, M.; Lou, Y.; Yang, X.F. DhhP, a Cyclic di-AMP Phosphodiesterase of Borrelia Burgdorferi, Is Essential for Cell Growth and Virulence. Infect. Immun. 2014, 82, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, F.; Liu, S.; Zhu, D.; Cong, H.; Gao, F.; Li, B.; Wang, H.; Lin, Z.; Liao, J.; et al. Structural and Biochemical Insight into the Mechanism of Rv2837c from Mycobacterium Tuberculosis as a C-di-NMP Phosphodiesterase. J. Biol. Chem. 2016, 291, 3668–3681. [Google Scholar] [CrossRef]
- Blötz, C.; Treffon, K.; Kaever, V.; Schwede, F.; Hammer, E.; Stülke, J. Identification of the Components Involved in Cyclic di-AMP Signaling in Mycoplasma Pneumoniae. Front. Microbiol. 2017, 8, 1328. [Google Scholar] [CrossRef]
- Manikandan, K.; Sabareesh, V.; Singh, N.; Saigal, K.; Mechold, U.; Sinha, K.M. Two-Step Synthesis and Hydrolysis of Cyclic di-AMP in Mycobacterium Tuberculosis. PLoS ONE 2014, 9, e86096. [Google Scholar] [CrossRef]
- Yang, J.; Bai, Y.; Zhang, Y.; Gabrielle, V.D.; Jin, L.; Bai, G. Deletion of the Cyclic di-AMP Phosphodiesterase Gene (cnpB) in Ycobacterium Tuberculosis Leads to Reduced Virulence in a Mouse Model of Infection. Mol. Microbiol. 2014, 93, 65–79. [Google Scholar] [CrossRef]
- Drexler, D.J.; Müller, M.; Rojas-Cordova, C.A.; Bandera, A.M.; Witte, G. Structural and Biophysical Analysis of the Soluble DHH/DHHA1-Type Phosphodiesterase TM1595 from Thermotoga maritima—ScienceDirect. Structure 2017, 25, 1887–1897. Available online: https://www.sciencedirect.com/science/article/pii/S0969212617303295?via%3Dihub (accessed on 31 January 2025).
- Dey, R.J.; Dey, B.; Zheng, Y.; Cheung, L.S.; Zhou, J.; Sayre, D.; Kumar, P.; Guo, H.; Lamichhane, G.; Sintim, H.O.; et al. Inhibition of Innate Immune Cytosolic Surveillance by an M. tuberculosis Phosphodiesterase. Nat. Chem. Biol. 2017, 13, 210–217. [Google Scholar] [CrossRef]
- Huynh, T.N.; Luo, S.; Pensinger, D.; Sauer, J.-D.; Tong, L.; Woodward, J.J. An HD-Domain Phosphodiesterase Mediates Cooperative Hydrolysis of c-di-AMP to Affect Bacterial Growth and Virulence. Proc. Natl. Acad. Sci. USA 2015, 112, E747–E756. [Google Scholar] [CrossRef]
- Bowman, L.; Zeden, M.S.; Schuster, C.F.; Kaever, V.; Gründling, A. New Insights into the Cyclic di-Adenosine Monophosphate (c-di-AMP) Degradation Pathway and the Requirement of the Cyclic Dinucleotide for Acid Stress Resistance in Staphylococcus aureus. J. Biol. Chem. 2016, 291, 26970–26986. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Aravind, L. Application of Comparative Genomics in the Identification and Analysis of Novel Families of Membrane-Associated Receptors in Bacteria. BMC Genomics 2003, 4, 34. [Google Scholar] [CrossRef]
- Karanja, C.W.; Yeboah, K.S.; Sintim, H.O. Identification of a Mycobacterium Tuberculosis Cyclic Dinucleotide Phosphodiesterase Inhibitor. ACS Infect. Dis. 2021, 7, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.R.; Sharma, S.; Kaadige, M.; Weston, A.; Thode, T. Phosphonates as Inhibitors of Enpp1 and Cdnp. WO2022125613A1, 16 June 2022. Available online: https://patents.google.com/patent/WO2022125613A1/en?oq=WO2022125613A1 (accessed on 2 February 2025).
- Rohilla, A.; Singh, A.K.; Koleske, B.; Srikrishna, G.; Bishai, W.R. Structure-Based Virtual Screening and In Vitro Validation of Inhibitors of Cyclic Dinucleotide Phosphodiesterases ENPP1 and CdnP. Microbiol. Spectr. 2023, 12, e02012-23. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular Function and Molecular Structure of Ecto-Nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Matange, N.; Podobnik, M.; Visweswariah, S.S. Metallophosphoesterases: Structural Fidelity with Functional Promiscuity. Biochem. J. 2015, 467, 201–216. [Google Scholar] [CrossRef]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are Essential Components of the IRF3 Signaling Pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Firon, A.; Dinis, M.; Raynal, B.; Poyart, C.; Trieu-Cuot, P.; Kaminski, P.A. Extracellular Nucleotide Catabolism by the Group B Streptococcus Ectonucleotidase NudP Increases Bacterial Survival in Blood. J. Biol. Chem. 2014, 289, 5479–5489. [Google Scholar] [CrossRef]
- Huynh, T.N.; Woodward, J.J. Too Much of a Good Thing: Regulated Depletion of c-di-AMP in the Bacterial Cytoplasm. Curr. Opin. Microbiol. 2016, 30, 22–29. [Google Scholar] [CrossRef]
- Rallu, F.; Gruss, A.; Ehrlich, S.D.; Maguin, E. Acid- and Multistress-Resistant Mutants of Lactococcus Lactis: Identification of Intracellular Stress Signals. Mol. Microbiol. 2000, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Helmann, J.D. Analysis of the Role of Bacillus Subtilis σM in β-Lactam Resistance Reveals an Essential Role for c-di-AMP in Peptidoglycan Homeostasis. Mol. Microbiol. 2012, 83, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Gretes, M.; Harlem, C.; Basuino, L.; Chambers, H.F. A mecA-Negative Strain of Methicillin-Resistant Staphylococcus aureus with High-Level β-Lactam Resistance Contains Mutations in Three Genes. Antimicrob. Agents Chemother. 2010, 54, 4900–4902. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.M.; O’Neill, A.J. Loss of Function of the GdpP Protein Leads to Joint β-Lactam/Glycopeptide Tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 579–581. [Google Scholar] [CrossRef]
- Uemura, Y.; Nakagawa, N.; Wakamatsu, T.; Kim, K.; Montelione, G.T.; Hunt, J.F.; Kuramitsu, S.; Masui, R. Crystal Structure of the Ligand-Binding Form of nanoRNase from Bacteroides fragilis, a Member of the DHH/DHHA1 Phosphoesterase Family of Proteins. FEBS Lett. 2013, 587, 2669–2674. [Google Scholar] [CrossRef]
- Wang, F.; He, Q.; Su, K.; Wei, T.; Xu, S.; Gu, L. Structural and Biochemical Characterization of the Catalytic Domains of GdpP Reveals a Unified Hydrolysis Mechanism for the DHH/DHHA1 Phosphodiesterase. Biochem. J. 2018, 475, 191–205. [Google Scholar] [CrossRef]
- Wang, X.; Davlieva, M.; Reyes, J.; Panesso, D.; Arias, C.A.; Shamoo, Y. A Novel Phosphodiesterase of the GdpP Family Modulates Cyclic di-AMP Levels in Response to Cell Membrane Stress in Daptomycin-Resistant Enterococci. Antimicrob. Agents Chemother. 2017, 61, 10-1128. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L. The Arms Race between Bacteria CBASS and Bacteriophages. Front. Immunol. 2023, 14, 1224341. [Google Scholar] [CrossRef]
- Hobbs, S.J.; Kranzusch, P.J. Nucleotide Immune Signaling in CBASS, Pycsar, Thoeris, and CRISPR Antiphage Defense. Annu. Rev. Microbiol. 2024, 78, 255–276. [Google Scholar] [CrossRef]
- Wang, J.Y.; Doudna, J.A. CRISPR Technology: A Decade of Genome Editing Is Only the Beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Athukoralage, J.S.; Rouillon, C.; Graham, S.; Grüschow, S.; White, M.F. Ring Nucleases Deactivate Type III CRISPR Ribonucleases by Degrading Cyclic Oligoadenylate. Nature 2018, 562, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Gauvin, C.C.; Charbonneau, A.A.; Burman, N.; Lawrence, C.M. Csx3 Is a Cyclic Oligonucleotide Phosphodiesterase Associated with Type III CRISPR–Cas That Degrades the Second Messenger cA4. J. Biol. Chem. 2020, 295, 14963–14972. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; Schwede, F.; Berk, C.; Rostøl, J.T.; Niewoehner, O.; Tejero, O.; Hall, J.; Marraffini, L.A.; Jinek, M. Activation and Self-Inactivation Mechanisms of the Cyclic Oligoadenylate-Dependent CRISPR Ribonuclease Csm6. Nat. Commun. 2020, 11, 1596. [Google Scholar] [CrossRef]
- Rohde, C.; Resch, G.; Pirnay, J.-P.; Blasdel, B.G.; Debarbieux, L.; Gelman, D.; Górski, A.; Hazan, R.; Huys, I.; Kakabadze, E.; et al. Expert Opinion on Three Phage Therapy Related Topics: Bacterial Phage Resistance, Phage Training and Prophages in Bacterial Production Strains. Viruses 2018, 10, 178. [Google Scholar] [CrossRef]
- Torres-Barceló, C. Phage Therapy Faces Evolutionary Challenges. Viruses 2018, 10, 323. [Google Scholar] [CrossRef]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically Aware Phage Therapy: Pharmacodynamic and Pharmacokinetic Obstacles to Phage Antibacterial Action in Animal and Human Bodies. Microbiol. Mol. Biol. Rev. 2019, 83, 10-1128. [Google Scholar] [CrossRef]
- Skurnik, M.; Alkalay-Oren, S.; Boon, M.; Clokie, M.; Sicheritz-Pontén, T.; Dąbrowska, K.; Hatfull, G.F.; Hazan, R.; Jalasvuori, M.; Kiljunen, S.; et al. Phage Therapy. Nat. Rev. Methods Primer 2025, 5, 9. [Google Scholar] [CrossRef]
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage Therapy: From Biological Mechanisms to Future Directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef]
- Athukoralage, J.S.; White, M.F. Cyclic Oligoadenylate Signaling and Regulation by Ring Nucleases during Type III CRISPR Defense. RNA 2021, 27, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Banh, D.V.; Roberts, C.G.; Morales-Amador, A.; Berryhill, B.A.; Chaudhry, W.; Levin, B.R.; Brady, S.F.; Marraffini, L.A. Bacterial cGAS Senses a Viral RNA to Initiate Immunity. Nature 2023, 623, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Richmond-Buccola, D.; Hobbs, S.J.; Garcia, J.M.; Toyoda, H.; Gao, J.; Shao, S.; Lee, A.S.Y.; Kranzusch, P.J. A Large-Scale Type I CBASS Antiphage Screen Identifies the Phage Prohead Protease as a Key Determinant of Immune Activation and Evasion. Cell Host Microbe 2024, 32, 1074–1088.e5. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Lau, R.K.; Mathews, I.T.; Birkholz, E.A.; Watrous, J.D.; Azimi, C.S.; Pogliano, J.; Jain, M.; Corbett, K.D. HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol. Cell 2020, 77, 709–722.e7. [Google Scholar] [CrossRef]
- Duncan-Lowey, B.; Kranzusch, P.J. CBASS Phage Defense and Evolution of Antiviral Nucleotide Signaling. Curr. Opin. Immunol. 2022, 74, 156–163. [Google Scholar] [CrossRef]
- Hobbs, S.J.; Wein, T.; Lu, A.; Morehouse, B.R.; Schnabel, J.; Leavitt, A.; Yirmiya, E.; Sorek, R.; Kranzusch, P.J. Phage Anti-CBASS and Anti-Pycsar Nucleases Subvert Bacterial Immunity. Nature 2022, 605, 522–526. [Google Scholar] [CrossRef]
- Cao, X.; Xiao, Y.; Huiting, E.; Cao, X.; Li, D.; Ren, J.; Fedorova, I.; Wang, H.; Guan, L.; Wang, Y.; et al. Phage Anti-CBASS Protein Simultaneously Sequesters Cyclic Trinucleotides and Dinucleotides. Mol. Cell 2024, 84, 375–385.e7. [Google Scholar] [CrossRef]
- Jia, N.; Jones, R.; Sukenick, G.; Patel, D.J. Second Messenger cA4 Formation within the Composite Csm1 Palm Pocket of Type III-A CRISPR-Cas Csm Complex and Its Release Path. Mol. Cell 2019, 75, 933–943.e6. [Google Scholar] [CrossRef]
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors—A STING in the Tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef]
- Riboscience, LLC. A First-in-Human, Phase 1 a/b Dose Escalation and Expansion Study to Evaluate RBS2418 as Monotherapy and in Combination with Pembrolizumab in Subjects with Advanced Unresectable, Recurrent or Metastatic Tumors. Clinical trial Registration NCT05270213; ClinicalTrials.gov; 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05270213 (accessed on 14 March 2023).
- Study Details | Evaluation of RBS2418 in Subjects with Advanced, Metastatic Solid Tumors | ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05270213 (accessed on 29 April 2024).
- Stingray Therapeutics. Phase 1, Dose Escalation, Safety, Tolerability, and Pharmacokinetic Study of SR-8541A (ENPP1 Inhibitor) Administered Orally as Monotherapy in Subjects with Advanced/Metastatic Solid Tumors. Clinical Trial Registration NCT06063681; ClinicalTrials.gov; 2023. Available online: https://clinicaltrials.gov/study/NCT06063681 (accessed on 20 February 2025).
- Txinno Bioscience Inc. A Multicenter, Open-Label, Phase 1 Dose Escalation and Expansion Study of TXN10128, an Inhibitor of ENPP1 in Subjects with Locally Advanced (Unresectable) or Metastatic Solid Tumors. Clinical Trial Registration NCT05978492; ClinicalTrials.gov; 2024. Available online: https://clinicaltrials.gov/study/NCT05978492 (accessed on 11 April 2025).
- Stingray Therapeutics. Phase 2 Study of SR-8541A in Combination with Botensilimab and Balstilimab in Subjects with Refractory Metastatic Microsatellite Stable Colorectal Cancer (MSS-CRC). Clinical Trial Registration NCT06589440; ClinicalTrials.gov; 2024. Available online: https://clinicaltrials.gov/study/NCT06589440 (accessed on 20 February 2025).
- Ruiz-Fernández de Córdoba, B.; Martínez-Monge, R.; Lecanda, F. ENPP1 Immunobiology as a Therapeutic Target. Clin. Cancer Res. 2023, 29, 2184–2193. [Google Scholar] [CrossRef]
- Huang, R.; Ning, Q.; Zhao, J.; Zhao, X.; Zeng, L.; Yi, Y.; Tang, S. Targeting ENPP1 for Cancer Immunotherapy: Killing Two Birds with One Stone. Biochem. Pharmacol. 2024, 220, 116006. [Google Scholar] [CrossRef]
- Guan, D.; Fang, L.; Feng, M.; Guo, S.; Xie, L.; Chen, C.; Sun, X.; Wu, Q.; Yuan, X.; Xie, Z.; et al. Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 Inhibitors: Research Progress and Prospects. Eur. J. Med. Chem. 2024, 267, 116211. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Ru, J.; Zhan, Z.; Lin, C.; Liu, Y.; Mao, W.; Zhang, J. Insight into Small-Molecule Inhibitors Targeting Extracellular Nucleotide Pyrophosphatase/Phosphodiesterase1 for Potential Multiple Human Diseases. Eur. J. Med. Chem. 2024, 268, 116286. [Google Scholar] [CrossRef] [PubMed]
- Solomon, P.E.; Bracken, C.J.; Carozza, J.A.; Wang, H.; Young, E.P.; Wellner, A.; Liu, C.C.; Sweet-Cordero, E.A.; Li, L.; Wells, J.A. Discovery of VH Domains That Allosterically Inhibit ENPP1. Nat. Chem. Biol. 2024, 20, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nishimasu, H.; Oikawa, D.; Hirano, S.; Hirano, H.; Kasuya, G.; Ishitani, R.; Tokunaga, F.; Nureki, O. Structural Insights into cGAMP Degradation by Ecto-Nucleotide Pyrophosphatase Phosphodiesterase 1. Nat. Commun. 2018, 9, 4424. [Google Scholar] [CrossRef]
- Carozza, J.A.; Brown, J.A.; Böhnert, V.; Fernandez, D.; AlSaif, Y.; Mardjuki, R.E.; Smith, M.; Li, L. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol. 2020, 27, 1347–1358.e5. [Google Scholar] [CrossRef]
- Hesketh, A.; Vergnano, M.; Wan, C.; Oliver, S.G. Bacterial Signaling Nucleotides Inhibit Yeast Cell Growth by Impacting Mitochondrial and Other Specifically Eukaryotic Functions. mBio 2017, 8, 10-1128. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vennard, C.S.; Oladeji, S.M.; Sintim, H.O. Inhibitors of Cyclic Dinucleotide Phosphodiesterases and Cyclic Oligonucleotide Ring Nucleases as Potential Drugs for Various Diseases. Cells 2025, 14, 663. https://doi.org/10.3390/cells14090663
Vennard CS, Oladeji SM, Sintim HO. Inhibitors of Cyclic Dinucleotide Phosphodiesterases and Cyclic Oligonucleotide Ring Nucleases as Potential Drugs for Various Diseases. Cells. 2025; 14(9):663. https://doi.org/10.3390/cells14090663
Chicago/Turabian StyleVennard, Christopher S., Samson Marvellous Oladeji, and Herman O. Sintim. 2025. "Inhibitors of Cyclic Dinucleotide Phosphodiesterases and Cyclic Oligonucleotide Ring Nucleases as Potential Drugs for Various Diseases" Cells 14, no. 9: 663. https://doi.org/10.3390/cells14090663
APA StyleVennard, C. S., Oladeji, S. M., & Sintim, H. O. (2025). Inhibitors of Cyclic Dinucleotide Phosphodiesterases and Cyclic Oligonucleotide Ring Nucleases as Potential Drugs for Various Diseases. Cells, 14(9), 663. https://doi.org/10.3390/cells14090663