
The Role of Oxidative Stress and Inflammation in the Pathogenesis and Treatment of Vascular Dementia




Abstract
1. Introduction
2. Vascular Dementia (VaD)
Definition and Etiology
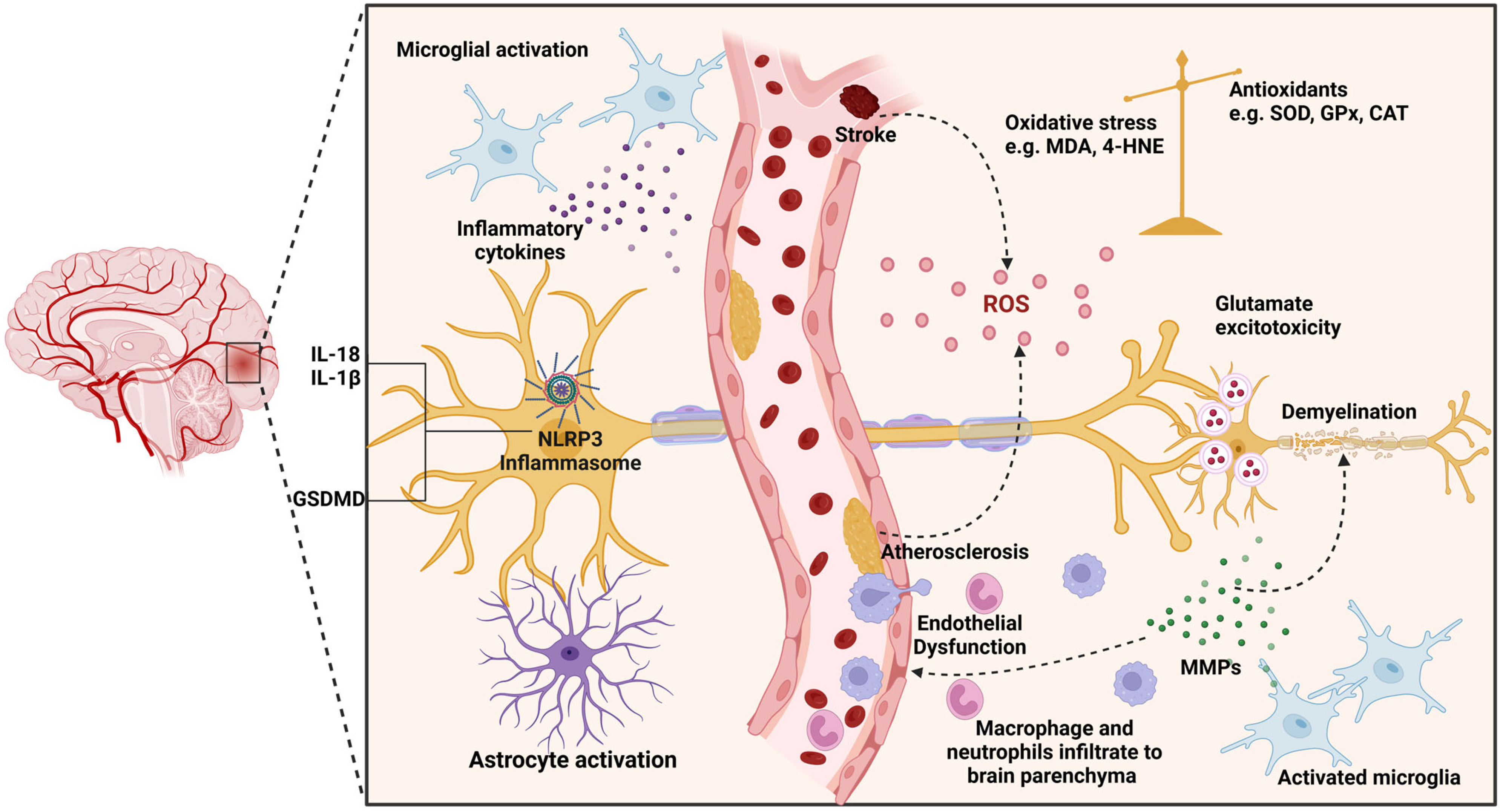
3. ROS Contribution to Neuronal Injury
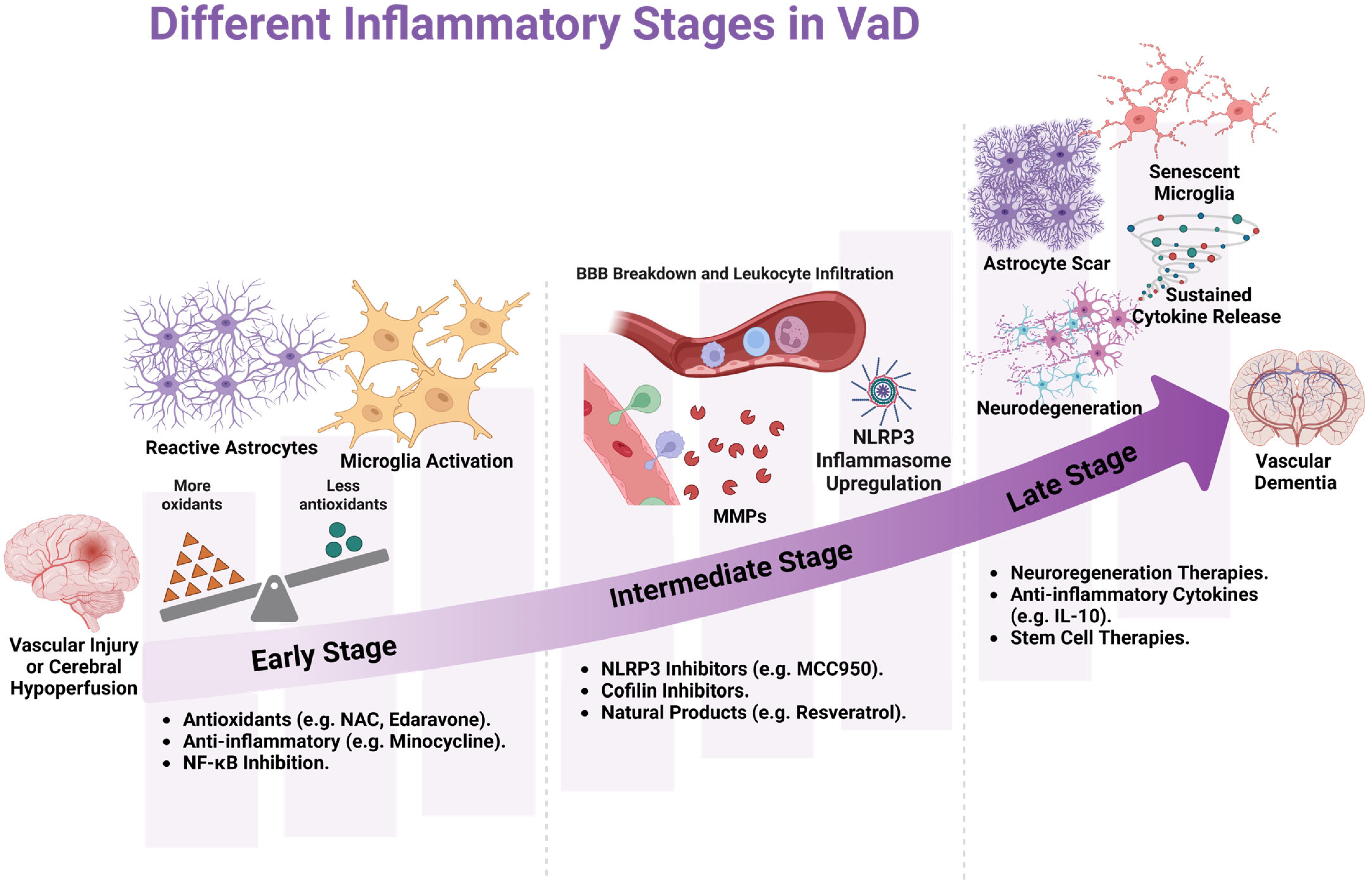
4. Neuroinflammation and VaD Progression
5. Animal Model to Induce VaD
6. Interventions Targeting Oxidative Stress and Inflammation in VaD
6.1. Use of Antioxidants and Anti-Inflammatory Agents in Preclinical Settings
6.2. Shortcomings of Anti-Inflammatories and Antioxidant Drugs
6.3. Use of Antioxidants and Anti-Inflammatory Agents in Clinical Settings

{kind=link}
{kind=link}
| Agent | Animal Model | Drug Protocol | Target/Pathway | Main Findings | Ref. |
|---|---|---|---|---|---|
| Banhabaekchulcheonma-Tang (BBCT) | BCAS, C57BL/6 mice | 80 mL/kg and 40 mL/kg PO, three times a week for 6 weeks starting 2 weeks after surgery | N/A | BBCT treatment significantly enhanced BCAS-associated memory impairment. It decreased microglia and astrocyte activation and reversed BCAS-dysregulated gene expression. BCCT exerts its neuroprotective effect by modulating neuropeptide signaling, promoting neuronal survival, and synaptic stability. | [111] |
| Berberine chloride | Permanent BCCAo, Wister rats | 50 mg/kg PO, daily for 2 months | N/A | Berberine administration increases spatial learning, mitigates memory impairment, alleviates histological damage, and suppresses AchE activity, improving cholinergic function. Additionally, it reduces apoptosis and necrosis in the CA1 region of the hippocampus. | [112] |
| Betulinic acid | Permanent BCCAo, Wistar rats | 10 and 15 mg/kg, PO, once daily starting from D8 to D30 after surgery | N/A | BA has a neuroprotective effect against memory impairment and neuroinflammation induced by VaD. It significantly improved cerebral blood flow after BCCAO reduced proinflammatory and oxidative stress markers and restored cAMP, cGMP, and neurotransmitters (e.g., DA, NE, and 5-HT3) to levels comparable to those in the sham group. | [113] |
| Bilobalide (Ginkgo biloba extract) | 2-VO, SD rats | 2, 4, and 8 mg/kg, IGAS for 2 months | N/A | BB treatment significantly alleviated memory and learning impairment associated with 2-VO. BB reduces oxidative stress (decreasing the MDA level and NOS activity and increasing the SOD activity and GSH level). It also mitigates neuronal histological changes associated with vascular insult in both the hippocampus and cortex. | [93] |
| Calmodulin inhibitor (DY-9836) | BCAS, C57BL/6 mice | 0.5 or 1 mg/kg, PO from D 5 to D 45 post-operation | NLRP3 inflammasome pathway | Repeated administration of DY-9836 attenuated BCAS-induced learning and cognitive impairment. It restored the phosphorylated CaMK-II in hippocampal CA1 neurons and reduced inflammation by inhibiting the NLRP3/Caspase-1/IL-1β signaling pathway. | [114] |
| Carnosine | BCCAO (alternate cycles of occlusion/relaxation of 10 min each for three cycles), Wistar rats | 200 or 400 mg/kg, IP once daily for 9 days after surgery | N/A | Carnosine administration mitigates CCH-induced spatial and cognitive impairment. It reduced oxidative stress by decreasing oxidative damage markers (e.g., AchE activity, MPO activity, and TBAR level) and increasing GSH levels. Its anti-inflammatory effect was observed by decreasing the neutrophil filtration. | [115] |
| Cilostazol and simvastatin | L-methionine-induced VaD, Wistar rats | Simvastatin (50 mg/kg, PO) or cilostazol (100 mg/kg, PO), respectively, for 32 days | N/A | Both drugs alleviate L-Met-induced memory impairment by decreasing oxidative stress and inflammation. Simvastatin and cilostazol attenuate AchE activity and increase brain endothelial nitric oxide synthase levels, reducing amyloid beta-42 and cholesterol levels. | [116,117] |
| Citicoline and Nicotinamide | Permanent BCCAO, SD rats | Citicoline (160 mg/kg) and Nicotinamide (40 mg/kg), IP once daily for 4 Weeks, started 1 week after surgery | SIRT1/TORC1/CREB pathway | Citicoline and NMN synergistically attenuate BCCAO-induced white matter damage and cognitive impairment by activating the SIRT1/TORC1/CREB pathway. They inhibit microglia activations, reduce proinflammatory cytokines (IL-1β, IL-6, and TNF-α), and increase anti-inflammatory mediators (e.g., IL-10). These findings highlight the neuroprotective effect of this combination. | [118] |
| Clostridium butyricum (probiotics) | permanent right unilateral common carotid artery occlusion (rUCCAO), ICR mice | (1 × 10⁶, 1 × 10⁷, and 1 × 10⁸ CFU/mL), 200 mcl IGAS daily for 6 weeks | BDNF-PI3K/Akt pathway | C. butyricum improved cognitive performance and decreased neuronal death in VaD by modulating the gut–brain axis and enhancing butyrate levels in the brain, ultimately activating the BDNF-PI3K/Akt signaling pathway. | [119] |
| Co-ultraPEALut (palmitoylethanolamide + luteolin) | BCCAO (alternate cycles of ligation/relaxation of 10 min each for three cycles), CD1 mice | 1 mg/kg, PO daily for 15 days | NF-κB | Co-ultraPEALut attenuates BCCAo-induced memory impairment. It inhibits the NF-κB activation by blocking IκB-α degradation, reducing proinflammatory markers (COX-2, iNOS), and decreasing oxidative stress (nitrotyrosine production). It inhibited apoptosis by decreasing Bax and increasing Bcl2 expression. It also exerts a neuroprotection effect on hippocampal neurons by increasing BDNF and NT-3 expression. | [120] |
| Danggui-Shaoyao San | Permanent BCCAo, SD rats | (1.8 g/kg or 7 g/kg) once daily for 4 weeks | IKK/NF-κB | DSS attenuated cognitive dysfunction in VaD rats induced by BCCAO, as observed by attenuating memory deficits, alleviating neuronal apoptosis through regulating the Bcl-2/Bax ratio, cleaved caspase-3, and oxidative stress pathways, and reducing oxidative stress markers (MDA, ROS). DSS has an anti-inflammatory effect, as shown by reducing TNF-α and IL-1β. | [121] |
| Danshen–Honghua Herbal Pair | Permanent BCCAO, Wistar rats | 3.2 g/kg/day, IGAS, for 4 weeks | N/A | DH enhanced cognitive performance in VaD rats. It also restored the cholinergic balance by increasing the Ach level and decreasing the AchE enzyme activity. It has a protective effect against neuronal apoptosis, as demonstrated by decreasing the ROS accumulation. | [122] |
| Dimethyl fumarate (DMF) | MCAO, SD rats | 12.5 mg/kg PO twice daily for 3 days before and 10 consecutive days after surgery | N/A | DMF treatment mitigated oxidative damage, oxidative stress, and neuroinflammation associated with post-stroke cognitive impairment. It improved cognitive function and reduced oxidative stress, apoptosis, and autophagosome formation in the hippocampal CA1 region. | [123] |
| Duloxetine | Permanent BCCAO, SD rats | 20 mg/kg, IP, once daily for 4 weeks | mTOR/S6K | DXT treatment protects against CCH-induced hippocampal neuronal damage in the CA1 region. Its neuroprotective effect is mediated by maintaining the TOR/S6K signaling pathway. DXT treatment also decreased proinflammatory biomarkers (e.g., TNF-α, IL-1β). | [124] |
| Edaravone | Permanent BCCAO, Wistar rats | 3, 5, and 6 mg/kg or IP for 28 days after surgery | ERK/Nrf2/HO-1 | Edaravone treatment significantly attenuates CCH-induced spatial and fear memory impairment. It reduces oxidative stress by enhancing the activity of antioxidants (e.g., SOD, HO-1) and decreasing the level of oxidative stress markers (e.g., MDA, LDH, ROS). Moreover, edaravone restored synaptic integrity by increasing the production of key hippocampal proteins and improving the phosphorylation of others critical for memory-related signals. | [125,126] |
| Edible bird’s nest (EBN) | Permanent BCCAO, SD rats | 60 mg/kg, 120 mg/kg PO, OD 8 weeks | N/A | Chronic EBN treatment attenuates CCH-induced cognitive impairment and pathological alteration in hippocampal neuronal cells. It preserved neuronal cell viability and reduced oxidative stress and neuroinflammation in the hippocampus, suggesting its potential as a neuroprotective drug to slow the progression of VaD. | [127] |
| Estrogen | Permanent BCCAO, SD rats | 17β-estradiol 100 µg/kg/day, IP for 8 weeks | Wnt/β-catenin pathway | Estrogen treatment significantly ameliorates cognitive damage and neuronal destruction. It inhibits autophagy by reducing the expression of Beclin-3 and LC3B. Furthermore, estrogen activates the Wnt/β-catenin pathway as indicated by increased expression of β-catenin and cyclin-D while decreasing the synthesis of glycogen kinase 3β. | [128] |
| Genistein | BCCAo (30 min), mice | 5.0 and 10 mg/kg PO, OD for 30 days | N/A | Genistein enhanced cognitive function, reduced neuronal apoptosis, and enhanced cellular viability in the CA1 region of the hippocampus. It also increased glucagon-like peptide-1 levels and inhibited dipeptidyl peptidase-4 activity. | [129] |
| GJ-4 | Permanent BCAS, mice | 50 mg/kg, IGAS, 4 weeks | Keap1-Nrf2/HO-1 pathway | GJ-4 improved both short- and long-term memory. It has a neuroprotective effect against VaD mediated by decreased oxidative stress through the Keap1-Nrf2/HO-1 pathway, enhancing lipid metabolism. Moreover, it has an antiapoptotic effect, seen by increasing the Bcl2/Bax ratio. GJ-4 also enhanced remyelination as indicated by increased expression of myelin-related protein (e.g., MBP, MOG, and MAG), which is essential for cognitive function improvement. | [130] |
| Ling-Yang-Gou-Teng-decoction (LG) | Autologous microthrombi injection against the background of hypercholesterolemia induced with a high fatty diet, Wistar rats | (2.58, 8.14, 25.80 g/ kg/day) PO, 3 days before and 3 weeks after microthrombi injection | N/A | Repeated LG exposure significantly enhanced cognitive function and memory ability. LG exerts its antioxidant effect by decreasing NOX2, a major source of oxidation in VaD, and upregulating the expression of SOD3. Moreover, LG mitigated the vascular and neural edema and increased neuronal hippocampus survival. | [131] |
| Melatonin | Permanent BCCAO, SD rats | 20 mcg/mL of melatonin in drinking water for 4 weeks | N/A | Chronic administration of the neurohormone melatonin alleviates VaD-associated neuronal damage by mitigating oxidative stress, illustrated by reducing oxidative stress markers (TBARS) and enhancing the antioxidant activity (SOD, CAT, GSH). Melatonin also decreases the apoptosis in the hippocampal CA1 neurons (modulates the Bcl-2/Bax ratio). | [132] |
| Osthole | Permanent BCCAo, SD rats | 5, 10, and 20 mg/kg PO daily for 62 days | NLRP3 inflammasome pathway | Chronic treatment of osthole alleviates cognitive impairment caused by BCCAo-induced VaD. It has an anti-inflammatory effect, indicated by decreasing microglial activation and downregulating NLRP3 pathway activation. Osthole also decreased Aβ deposition and reduced the expression of APP and BACE1. | [133] |
| Paeoniflorin | 4-VO, SD rats | 40 mg/kg IP once daily for 4 weeks | mTOR/NF-κB and PI3K/Akt pathways | PF mitigated cognitive impairment, decreased proinflammatory biomarkers (IL-1β, IL-6, and TNF-α), and increased anti-inflammatory markers (e.g., IL-10 and TGF-β) in the hippocampus. It also protected against morphological damage of hippocampal neurons by activating the PI3K/Akt pathway, which shifts microglial polarization to the M2 phenotype. | [134] |
| 20 (S)—Protopanaxadiol (PPD) | Permanent BCCAo, SD rats | PPD- H, M, L; 20, 10, 5 mg/kg, respectively, SQ, once daily for 3 weeks | NLRP3 inflammasome pathway | PPD exerts neuroprotective effects in VaD rats mainly due to its anti-inflammatory effect, which is mediated by reducing NLRP3 inflammasome activation, preventing amyloid-beta precipitation, and tau phosphorylation. | [67] |
| Resveratrol | Permanent BCCAO, Wistar rats | 25 mg/kg PO, daily for 4 weeks from W8 to W12 after surgery | N/A | Resveratrol ameliorates CCH-induced spatial memory impairment. It significantly attenuates the progression of VaD by reducing the apoptosis, as indicated by decreasing the Bax/Bcl2 ratio and reducing the c-caspase3 and c-PARP protein expression. | [135] |
| Resveratrol-loaded solid lipid nanoparticles (R-SLNs) | Permanent BCCAO, SD rats | 10 mg/kg, PO once daily, starting from W4 to W8 after surgery | Nrf2/HO-1 pathway | R-SLN supplementation provided a neuroprotective effect against VaD. It improved spatial memory retention, mitigated CCH-induced oxidative stress in brain tissue as demonstrated by decreased levels of oxidative stress markers (e.g., MDA, lipid peroxidation, protein carbonyl, and GSSG), and increased the level/activity of antioxidants (HO-1, NRF-2, SOD, and GSH). | [136] |
| Tetrandrine | 2-VO, SD rats | 10 mg/kg or 30 mg/kg, IP, Q.o.d for 4 weeks | N/A | Tetrandrine enhanced cognitive performance and attenuated 2-VO-associated hippocampal neuronal necrosis. It decreased IL-1β levels and reduced NR2B phosphorylation. | [137] |
| Vildagliptin | Pancreatectomy-induced VaD, Wistar rats | 3 and 6 mg/kg PO, daily for 2 months | Chronic vildagliptin administration significantly attenuated diabetes-induced memory and executive functioning impairment. Vildagliptin also restored endothelial function and reduced oxidative stress (decreased TBARS and MPO and increased GSH). Moreover, vildagliptin improved BBB integrity and reduced brain calcium levels. | [138] | |
| Zafirlukast, piracetam, and their combination | L-Met-induced VaD, Wistar rats | Zafirlukast (20 mg/kg, PO), piracetam (600 mg/kg, PO), or combination (zafirlukast 20 mg/kg + piracetam 600 mg/kg, PO) OD for 32 days | N/A | Both agents, alone and in combination, improved the behavioral and neurochemical alteration associated with L-Met administration. Zafirlukast and piracetam decreased norepinephrine, dopamine, and Aβ-42 levels; increased Ach levels; and decreased AchE activity. Both drugs enhanced GSH and IL-10 content and reduced IL-6 and MDA levels. | [139] |
7. The Future of Antioxidants and Anti-Inflammatories as a Therapeutic Target in VaD
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chang Wong, E.; Chang Chui, H. Vascular Cognitive Impairment and Dementia. Continuum 2022, 28, 750–780. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Ikram, M.A. Epidemiology of vascular dementia: Nosology in a time of epiomics. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Zhang, B.; Xia, R.; Jia, Q.Y. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9601–9614. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef]
- Linh, T.T.D.; Hsieh, Y.C.; Huang, L.K.; Hu, C.J. Clinical Trials of New Drugs for Vascular Cognitive Impairment and Vascular Dementia. Int. J. Mol. Sci. 2022, 23, 11067. [Google Scholar] [CrossRef]
- Rundek, T.; Tolea, M.; Ariko, T.; Fagerli, E.A.; Camargo, C.J. Vascular Cognitive Impairment (VCI). Neurotherapeutics 2022, 19, 68–88. [Google Scholar] [CrossRef]
- Cervellati, C.; Romani, A.; Seripa, D.; Cremonini, E.; Bosi, C.; Magon, S.; Passaro, A.; Bergamini, C.M.; Pilotto, A.; Zuliani, G. Oxidative balance, homocysteine, and uric acid levels in older patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J. Neurol. Sci. 2014, 337, 156–161. [Google Scholar] [CrossRef]
- Luca, M.; Luca, A.; Calandra, C. The Role of Oxidative Damage in the Pathogenesis and Progression of Alzheimer’s Disease and Vascular Dementia. Oxidative Med. Cell. Longev. 2015, 2015, 504678. [Google Scholar] [CrossRef]
- Tian, Z.; Ji, X.; Liu, J. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. Int. J. Mol. Sci. 2022, 23, 6224. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; Van Swieten, J.C.; Stijnen, T.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Inflammatory proteins in plasma and the risk of dementia: The rotterdam study. Arch. Neurol. 2004, 61, 668–672. [Google Scholar] [CrossRef]
- Gervois, P.; Lambrichts, I. The emerging role of triggering receptor expressed on myeloid cells 2 as a target for immunomodulation in ischemic stroke. Front. Immunol. 2019, 10, 1668. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Khan, M.W.; Javalkar, V.; Toledo, E.G.; Kelley, R.E. Emerging Concepts in Vascular Dementia: A Review. J. Stroke Cerebrovasc. Dis. 2021, 30, 105864. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Skrobot, O.A.; O’Brien, J.; Black, S.; Chen, C.; DeCarli, C.; Erkinjuntti, T.; Ford, G.A.; Kalaria, R.N.; Pantoni, L.; Pasquier, F.; et al. The Vascular Impairment of Cognition Classification Consensus Study. Alzheimer’s Dement. 2017, 13, 624–633. [Google Scholar] [CrossRef]
- Kang, P.; Ying, C.; Chen, Y.; Ford, A.L.; An, H.; Lee, J.-M. Oxygen metabolic stress and white matter injury in patients with cerebral small vessel disease. Stroke 2022, 53, 1570–1579. [Google Scholar] [CrossRef]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef]
- Inoue, Y.; Shue, F.; Bu, G.; Kanekiyo, T. Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 46. [Google Scholar] [CrossRef]
- Lecordier, S.; Manrique-Castano, D.; El Moghrabi, Y.; ElAli, A. Neurovascular Alterations in Vascular Dementia: Emphasis on Risk Factors. Front. Aging Neurosci. 2021, 13, 727590. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, Y.; Zhao, Y.; Huang, T.Y.; Zheng, Q.; Wang, X. Peripheral and central neuroimmune mechanisms in Alzheimer’s disease pathogenesis. Mol. Neurodegener. 2025, 20, 22. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Singh, S.; Sharma, A. Role of Reactive Oxygen Species (ROS) and Cytokines in Vascular Dementia: A Review. J. Pharm. Teach. 2015, 6, 2620–2629. [Google Scholar]
- Zhao, H.; Zhang, R.; Yan, X.; Fan, K. Superoxide dismutase nanozymes: An emerging star for anti-oxidation. J. Mater. Chem. B 2021, 9, 6939–6957. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.J.; Boling, W.; Kindy, M.S.; Peschon, J.; Kraemer, P.J.; Carpenter, M.K.; Holtsberg, F.W.; Mattson, M.P. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996, 2, 788–794. [Google Scholar] [CrossRef]
- Han, B.; Jiang, W.; Liu, H.; Wang, J.; Zheng, K.; Cui, P.; Feng, Y.; Dang, C.; Bu, Y.; Wang, Q.M. Upregulation of neuronal PGC-1α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 2020, 10, 2832. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef]
- Chen, Q.; Ding, Q.; Thorpe, J.; Dohmen, R.J.; Keller, J.N. RNA interference toward UMP1 induces proteasome inhibition in Saccharomyces cerevisiae: Evidence for protein oxidation and autophagic cell death. Free Radic. Biol. Med. 2005, 38, 226–234. [Google Scholar] [CrossRef]
- Christophe, M.; Nicolas, S. Mitochondria: A target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr. Pharm. Des. 2006, 12, 739–757. [Google Scholar] [CrossRef]
- Rajeev, V.; Fann, D.Y.; Dinh, Q.N.; Kim, H.A.; De Silva, T.M.; Lai, M.K.P.; Chen, C.L.; Drummond, G.R.; Sobey, C.G.; Arumugam, T.V. Pathophysiology of blood brain barrier dysfunction during chronic cerebral hypoperfusion in vascular cognitive impairment. Theranostics 2022, 12, 1639–1658. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Chiba, Y.; Matsumoto, K.; Murakami, R.; Fujihara, R.; Kawauchi, M.; Miyanaka, H.; Nakagawa, T. Blood-brain barrier damage in vascular dementia. Neuropathology 2016, 36, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mattioli, P.; Aldred, S.; Cecchetti, R.; Stahl, W.; Griffiths, H.; Senin, U.; Sies, H.; Mecocci, P. Plasma antioxidant status, immunoglobulin g oxidation and lipid peroxidation in demented patients: Relevance to Alzheimer disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 2004, 18, 265–270. [Google Scholar] [CrossRef]
- Casado, Á.; Encarnación López-Fernández, M.; Concepción Casado, M.; de La Torre, R. Lipid Peroxidation and Antioxidant Enzyme Activities in Vascular and Alzheimer Dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef]
- Bednarz-Misa, I.; Berdowska, I.; Zboch, M.; Misiak, B.; Zieliński, B.; Płaczkowska, S.; Fleszar, M.; Wiśniewski, J.; Gamian, A.; Krzystek-Korpacka, M. Paraoxonase 1 decline and lipid peroxidation rise reflect a degree of brain atrophy and vascular impairment in dementia. Adv. Clin. Exp. Med. 2020, 29, 71–78. [Google Scholar] [CrossRef]
- Gustaw-Rothenberg, K.; Kowalczuk, K.; Stryjecka-Zimmer, M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatr. Gerontol. Int. 2010, 10, 161–166. [Google Scholar] [CrossRef]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.-M.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef]
- Suridjan, I.; Herrmann, N.; Adibfar, A.; Saleem, M.; Andreazza, A.; Oh, P.I.; Lanctôt, K.L. Lipid Peroxidation Markers in Coronary Artery Disease Patients with Possible Vascular Mild Cognitive Impairment. J. Alzheimer’s Dis. 2017, 58, 885–896. [Google Scholar] [CrossRef]
- Sun, A.Y.; Chen, Y.-M. Oxidative stress and neurodegenerative disorders. J. Biomed. Sci. 1998, 5, 401–414. [Google Scholar] [CrossRef]
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C.; et al. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Milatovic, D.; Zaja-Milatovic, S.; Breyer, R.M.; Aschner, M.; Montine, T.J. Chapter 55—Neuroinflammation and Oxidative Injury in Developmental Neurotoxicity. In Reproductive and Developmental Toxicology, 2nd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1051–1061. [Google Scholar] [CrossRef]
- Milatovic, D.; Zaja-Milatovic, S.; Breyer, R.M.; Aschner, M.; Montine, T.J. Chapter 64—Neuroinflammation and oxidative injury in developmental neurotoxicity. In Reproductive and Developmental Toxicology; Gupta, R.C., Ed.; Academic Press: San Diego, CA, USA, 2011; pp. 847–854. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S369–S379. [Google Scholar] [CrossRef]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An integrating overview of reactive-Neuroimmune cell interactions in health and disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Lee, K.M.; Bang, J.; Kim, B.Y.; Lee, I.S.; Han, J.S.; Hwang, B.Y.; Jeon, W.K. Fructus mume alleviates chronic cerebral hypoperfusion-induced white matter and hippocampal damage via inhibition of inflammation and downregulation of TLR4 and p38 MAPK signaling. BMC Complement. Altern. Med. 2015, 15, 125. [Google Scholar] [CrossRef]
- Park, J.-H.; Seo, Y.H.; Jang, J.-H.; Jeong, C.-H.; Lee, S.; Park, B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. J. Neuroinflamm. 2017, 14, 240. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar] [CrossRef]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Uslu, S.; Akarkarasu, Z.E.; Ozbabalik, D.; Ozkan, S.; Colak, O.; Demirkan, E.S.; Ozkiris, A.; Demirustu, C.; Alatas, O. Levels of amyloid beta-42, interleukin-6 and tumor necrosis factor-alpha in Alzheimer’s disease and vascular dementia. Neurochem. Res. 2012, 37, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Custodero, C.; Ciavarella, A.; Panza, F.; Gnocchi, D.; Lenato, G.M.; Lee, J.; Mazzocca, A.; Sabbà, C.; Solfrizzi, V. Role of inflammatory markers in the diagnosis of vascular contributions to cognitive impairment and dementia: A systematic review and meta-analysis. Geroscience 2022, 44, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Poh, L.; Sim, W.L.; Jo, D.G.; Dinh, Q.N.; Drummond, G.R.; Sobey, C.G.; Chen, C.L.; Lai, M.K.P.; Fann, D.Y.; Arumugam, T.V. The role of inflammasomes in vascular cognitive impairment. Mol. Neurodegener. 2022, 17, 4. [Google Scholar] [CrossRef]
- Liu, Q.; He, S.; Groysman, L.; Shaked, D.; Russin, J.; Cen, S.; Mack, W.J. White matter injury due to experimental chronic cerebral hypoperfusion is associated with C5 deposition. PLoS ONE 2013, 8, e84802. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Mossanen Parsi, M.; Duval, C.; Ariëns, R.A.S. Vascular Dementia and Crosstalk Between the Complement and Coagulation Systems. Front. Cardiovasc. Med. 2021, 8, 803169. [Google Scholar] [CrossRef]
- Shinjyo, N.; Kagaya, W.; Pekna, M. Interaction Between the Complement System and Infectious Agents—A Potential Mechanistic Link to Neurodegeneration and Dementia. Front. Cell. Neurosci. 2021, 15, 710390. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 425–437. [Google Scholar] [CrossRef]
- Bjerke, M.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A.; Sullivan, N.; Esiri, M.M. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke 2001, 32, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Chopp, M.; Chen, J. Models and mechanisms of vascular dementia. Exp. Neurol. 2015, 272, 97–108. [Google Scholar] [CrossRef]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.; Rosenberg, G.A. Rodent Models of Vascular Cognitive Impairment. Transl. Stroke Res. 2016, 7, 407–414. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Y.-j.; Niu, T.-y.; Chen, T.-t.; Li, H.-b.; Wu, S.-h.; Li, G.-l. Neuroprotective effect of 20 (S)—Protopanaxadiol (PPD) attenuates NLRP3 inflammasome-mediated microglial pyroptosis in vascular dementia rats. Neurosci. Lett. 2023, 814, 137439. [Google Scholar] [CrossRef]
- Poh, L.; Fann, D.Y.; Wong, P.; Lim, H.M.; Foo, S.L.; Kang, S.W.; Rajeev, V.; Selvaraji, S.; Iyer, V.R.; Parathy, N.; et al. AIM2 inflammasome mediates hallmark neuropathological alterations and cognitive impairment in a mouse model of vascular dementia. Mol. Psychiatry 2021, 26, 4544–4560. [Google Scholar] [CrossRef]
- Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20, 6176. [Google Scholar] [CrossRef]
- Chiang, T.; Messing, R.O.; Chou, W.H. Mouse model of middle cerebral artery occlusion. J. Vis. Exp. 2011, 2761. [Google Scholar] [CrossRef]
- Wang, C.; Ma, Z.; Wang, Z.; Ming, S.; Ding, Y.; Zhou, S.; Qian, H. Eriodictyol Attenuates MCAO-Induced Brain Injury and Neurological Deficits via Reversing the Autophagy Dysfunction. Front. Syst. Neurosci. 2021, 15, 655125. [Google Scholar] [CrossRef]
- Johnson, A.C.; Miller, J.E.; Cipolla, M.J. Memory impairment in spontaneously hypertensive rats is associated with hippocampal hypoperfusion and hippocampal vascular dysfunction. J. Cereb. Blood Flow Metab. 2020, 40, 845–859. [Google Scholar] [CrossRef]
- Roberts, R.; Huckstepp, R.T.R. Innate Sleep Apnea in Spontaneously Hypertensive Rats Is Associated With Microvascular Rarefaction and Neuronal Loss in the preBötzinger Complex. Stroke 2023, 54, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, M.; Catalani, A.; Consoli, C.; Marletta, N.; Tomassoni, D.; Avola, R. The hippocampus in spontaneously hypertensive rats: An animal model of vascular dementia? Mech. Ageing Dev. 2002, 123, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.B.; Jonak, E. Cerebrovascular disease and dementia: A primate model of hypertension and cognition. Alzheimer’s Dement. 2007, 3, S6–S15. [Google Scholar] [CrossRef] [PubMed]
- Dmytriv, T.R.; Duve, K.V.; Storey, K.B.; Lushchak, V.I. Vicious cycle of oxidative stress and neuroinflammation in pathophysiology of chronic vascular encephalopathy. Front. Physiol. 2024, 15, 1443604. [Google Scholar] [CrossRef]
- Chang, D.; Liu, J.; Bilinski, K.; Xu, L.; Steiner, G.Z.; Seto, S.W.; Bensoussan, A. Herbal Medicine for the Treatment of Vascular Dementia: An Overview of Scientific Evidence. Evid. Based Complement. Altern. Med. 2016, 2016, 7293626. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Mather, A.E.; Peters, R.; Lawrence, C.B.; Brough, D. Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun. 2020, 2, fcaa109. [Google Scholar] [CrossRef]
- Sastre, M.; Gentleman, S.M. NSAIDs: How They Work and Their Prospects as Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2010, 2, 20. [Google Scholar] [CrossRef]
- Poljsak, B. Strategies for reducing or preventing the generation of oxidative stress. Oxidative Med. Cell. Longev. 2011, 2011, 194586. [Google Scholar] [CrossRef]
- Spence, J.D.; Grosser, T.; FitzGerald, G.A. Acetaminophen, Nonsteroidal Anti-Inflammatory Drugs, and Hypertension. Hypertension 2022, 79, 1922–1926. [Google Scholar] [CrossRef]
- Tai, F.W.D.; McAlindon, M.E. Non-steroidal anti-inflammatory drugs and the gastrointestinal tract. Clin. Med. 2021, 21, 131–134. [Google Scholar] [CrossRef]
- Li, S.; Fasipe, B.; Laher, I. Potential harms of supplementation with high doses of antioxidants in athletes. J. Exerc. Sci. Fit. 2022, 20, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Labriola, D.; Livingston, R. Possible interactions between dietary antioxidants and chemotherapy. Oncology 1999, 13, 1003–1008, discussion 1008, 1011–1002. [Google Scholar] [PubMed]
- Wieland, L.S.; Moffet, I.; Shade, S.; Emadi, A.; Knott, C.; Gorman, E.F.; D’Adamo, C. Risks and benefits of antioxidant dietary supplement use during cancer treatment: Protocol for a scoping review. BMJ Open 2021, 11, e047200. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.; Pollack, C.; Butkerait, P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther. Clin. Risk Manag. 2015, 11, 1061–1075. [Google Scholar] [CrossRef]
- Oken, B.S.; Storzbach, D.M.; Kaye, J.A. The Efficacy of Ginkgo biloba on Cognitive Function in Alzheimer Disease. Arch. Neurol. 1998, 55, 1409–1415. [Google Scholar] [CrossRef]
- Pagotto, G.L.O.; Santos, L.; Osman, N.; Lamas, C.B.; Laurindo, L.F.; Pomini, K.T.; Guissoni, L.M.; Lima, E.P.; Goulart, R.A.; Catharin, V.; et al. Ginkgo biloba: A Leaf of Hope in the Fight against Alzheimer’s Dementia: Clinical Trial Systematic Review. Antioxidants 2024, 13, 651. [Google Scholar] [CrossRef]
- Wąsik, A.; Antkiewicz-Michaluk, L. The mechanism of neuroprotective action of natural compounds. Pharmacol. Rep. 2017, 69, 851–860. [Google Scholar] [CrossRef]
- von Gunten, A.; Schlaefke, S.; Überla, K. Efficacy of Ginkgo biloba extract EGb 761® in dementia with behavioural and psychological symptoms: A systematic review. World J. Biol. Psychiatry 2016, 17, 622–633. [Google Scholar] [CrossRef]
- García-Alberca, J.M.; Mendoza, S.; Gris, E. Benefits of Treatment with Ginkgo Biloba Extract EGb 761 Alone or Combined with Acetylcholinesterase Inhibitors in Vascular Dementia. Clin. Drug Investig. 2022, 42, 391–402. [Google Scholar] [CrossRef]
- Morató, X.; Marquié, M.; Tartari, J.P.; Lafuente, A.; Abdelnour, C.; Alegret, M.; Jofresa, S.; Buendía, M.; Pancho, A.; Aguilera, N.; et al. A randomized, open-label clinical trial in mild cognitive impairment with EGb 761 examining blood markers of inflammation and oxidative stress. Sci. Rep. 2023, 13, 5406. [Google Scholar] [CrossRef]
- Li, W.Z.; Wu, W.Y.; Huang, H.; Wu, Y.Y.; Yin, Y.Y. Protective effect of bilobalide on learning and memory impairment in rats with vascular dementia. Mol. Med. Rep. 2013, 8, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rojas, B.; Gómez-Sierra, T.; Medina-Campos, O.N.; Hernández-Juárez, J.; Hernández-Cruz, P.A.; Gallegos-Velasco, I.B.; Pérez-Cervera, Y.; Pedraza-Chaverri, J. Antioxidant activity of glucosamine and its effects on ROS production, Nrf2, and O-GlcNAc expression in HMEC-1 cells. Curr. Res. Toxicol. 2023, 5, 100128. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Lampe, J.W.; Vaughan, T.L.; Peters, U.; Rehm, C.D.; White, E. Association between use of specialty dietary supplements and C-reactive protein concentrations. Am. J. Epidemiol. 2012, 176, 1002–1013. [Google Scholar] [CrossRef]
- Binsi, P.K.; Zynudheen, A.A. 4—Functional and Nutraceutical Ingredients From Marine Resources. In Value-Added Ingredients and Enrichments of Beverages; Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 101–171. [Google Scholar] [CrossRef]
- Nevado-Holgado, A.J.; Kim, C.H.; Winchester, L.; Gallacher, J.; Lovestone, S. Commonly prescribed drugs associate with cognitive function: A cross-sectional study in UK Biobank. BMJ Open 2016, 6, e012177. [Google Scholar] [CrossRef]
- Zheng, J.; Ni, C.; Zhang, Y.; Huang, J.; Hukportie, D.N.; Liang, B.; Tang, S. Association of regular glucosamine use with incident dementia: Evidence from a longitudinal cohort and Mendelian randomization study. BMC Med. 2023, 21, 114. [Google Scholar] [CrossRef]
- Wang, A.; Jia, B.; Zhang, X.; Huo, X.; Chen, J.; Gui, L.; Cai, Y.; Guo, Z.; Han, Y.; Peng, Z.; et al. Efficacy and Safety of Butylphthalide in Patients With Acute Ischemic Stroke: A Randomized Clinical Trial. JAMA Neurol. 2023, 80, 851–859. [Google Scholar] [CrossRef]
- Rashid, M.H.; Babu, D.; Siraki, A.G. Interactions of the antioxidant enzymes NAD(P)H: Quinone oxidoreductase 1 (NQO1) and NRH: Quinone oxidoreductase 2 (NQO2) with pharmacological agents, endogenous biochemicals and environmental contaminants. Chem. Biol. Interact. 2021, 345, 109574. [Google Scholar] [CrossRef]
- Qi, F.X.; Hu, Y.; Kang, L.J.; Li, P.; Gao, T.C.; Zhang, X. Effects of Butyphthalide Combined with Idebenone on Inflammatory Cytokines and Vascular Endothelial Functions of Patients with Vascular Dementia. J. Coll. Physicians Surg. Pak. 2020, 30, 23–27. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Qi, X.; Wu, F.; Zhang, D. Effect of butylphthalide combined with idebenone on vascular dementia: A retrospective observational analysis. Medicine 2024, 103, e37495. [Google Scholar] [CrossRef]
- Bath, P.M.; Mhlanga, I.; Woodhouse, L.J.; Doubal, F.; Oatey, K.; Montgomery, A.A.; Wardlaw, J.M. Cilostazol and isosorbide mononitrate for the prevention of progression of cerebral small vessel disease: Baseline data and statistical analysis plan for the Lacunar Intervention Trial-2 (LACI-2) (ISRCTN14911850). Stroke Vasc. Neurol. 2023, 8, 134–143. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Woodhouse, L.J.; Mhlanga, I.I.; Oatey, K.; Heye, A.K.; Bamford, J.; Cvoro, V.; Doubal, F.N.; England, T.; Hassan, A.; et al. Isosorbide Mononitrate and Cilostazol Treatment in Patients With Symptomatic Cerebral Small Vessel Disease: The Lacunar Intervention Trial-2 (LACI-2) Randomized Clinical Trial. JAMA Neurol. 2023, 80, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Blair, G.W.; Appleton, J.P.; Law, Z.K.; Doubal, F.; Flaherty, K.; Dooley, R.; Shuler, K.; Richardson, C.; Hamilton, I.; Shi, Y.; et al. Preventing cognitive decline and dementia from cerebral small vessel disease: The LACI-1 Trial. Protocol and statistical analysis plan of a phase IIa dose escalation trial testing tolerability, safety and effect on intermediary endpoints of isosorbide mononitrate and cilostazol, separately and in combination. Int. J. Stroke 2018, 13, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H.; Kim, G.W.; Lee, Y.R.; Park, D.K.; Song, B.; Kim, D.S. Effects of Sildenafil on Cognitive Function Recovery and Neuronal Cell Death Protection After Transient Global Cerebral Ischemia in Gerbils. Biomedicines 2024, 12, 2077. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Pérez-Roldán, J.M.; García-Barroso, C.; Franco, R.; Aguirre, N.; García-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef]
- Xiong, Y.; Wintermark, P. The Role of Sildenafil in Treating Brain Injuries in Adults and Neonates. Front. Cell. Neurosci. 2022, 16, 879649. [Google Scholar] [CrossRef]
- Yu, Y.H.; Kim, S.W.; Kang, J.; Song, Y.; Im, H.; Kim, S.J.; Yoo, D.Y.; Lee, M.R.; Park, D.K.; Oh, J.S.; et al. Phosphodiesterase-5 Inhibitor Attenuates Anxious Phenotypes and Movement Disorder Induced by Mild Ischemic Stroke in Rats. J. Korean Neurosurg. Soc. 2022, 65, 665–679. [Google Scholar] [CrossRef]
- Webb, A.; Werring, D.; Dawson, J.; Rothman, A.; Lawson, A.; Wartolowska, K. Design of a randomised, double-blind, crossover, placebo-controlled trial of effects of sildenafil on cerebrovascular function in small vessel disease: Oxford haemodynamic adaptation to reduce pulsatility trial (OxHARP). Eur. Stroke J. 2021, 6, 283–290. [Google Scholar] [CrossRef]
- Kim, D.W.; Lim, J.H.; Cho, S.; Kim, S.H. Effects of Banhabaekchulcheonma-Tang on Brain Injury and Cognitive Function Impairment Caused by Bilateral Common Carotid Artery Stenosis in a Mouse Model. Int. J. Med. Sci. 2024, 21, 644–655. [Google Scholar] [CrossRef]
- Aski, M.L.; Rezvani, M.E.; Khaksari, M.; Hafizi, Z.; Pirmoradi, Z.; Niknazar, S.; Mehrjerdi, F.Z. Neuroprotective effect of berberine chloride on cognitive impairment and hippocampal damage in experimental model of vascular dementia. Iran. J. Basic Med. Sci. 2018, 21, 53–58. [Google Scholar] [CrossRef]
- Kaundal, M.; Zameer, S.; Najmi, A.K.; Parvez, S.; Akhtar, M. Betulinic acid, a natural PDE inhibitor restores hippocampal cAMP/cGMP and BDNF, improve cerebral blood flow and recover memory deficits in permanent BCCAO induced vascular dementia in rats. Eur. J. Pharmacol. 2018, 832, 56–66. [Google Scholar] [CrossRef]
- Wang, R.; Yin, Y.X.; Mahmood, Q.; Wang, X.J.; Gao, Y.P.; Gou, G.J.; Ahmed, M.M.; Kohji, F.; Du, Y.Z.; Han, F. Calmodulin inhibitor ameliorates cognitive dysfunction via inhibiting nitrosative stress and NLRP3 signaling in mice with bilateral carotid artery stenosis. CNS Neurosci. Ther. 2017, 23, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Bhatia, P.; Kumar, A.; Jaggi, A.S.; Singh, N. Potential of carnosine, a histamine precursor in rat model of bilateral common carotid artery occlusion-induced vascular dementia. Fundam. Clin. Pharmacol. 2018, 32, 516–531. [Google Scholar] [CrossRef] [PubMed]
- El-Dessouki, A.M.; Galal, M.A.; Awad, A.S.; Zaki, H.F. Neuroprotective Effects of Simvastatin and Cilostazol in l-Methionine-Induced Vascular Dementia in Rats. Mol. Neurobiol. 2017, 54, 5074–5084. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Trigiani, L.J.; Hamel, E. High cholesterol triggers white matter alterations and cognitive deficits in a mouse model of cerebrovascular disease: Benefits of simvastatin. Cell Death Dis. 2019, 10, 89. [Google Scholar] [CrossRef]
- Zhao, N.; Zhu, X.; Xie, L.; Guan, X.; Tang, L.; Jiang, G.; Pang, T. The Combination of Citicoline and Nicotinamide Mononucleotide Induces Neurite Outgrowth and Mitigates Vascular Cognitive Impairment via SIRT1/CREB Pathway. Cell. Mol. Neurobiol. 2023, 43, 4261–4277. [Google Scholar] [CrossRef]
- Liu, J.; Sun, J.; Wang, F.; Yu, X.; Ling, Z.; Li, H.; Zhang, H.; Jin, J.; Chen, W.; Pang, M.; et al. Neuroprotective Effects of Clostridium butyricum Against Vascular Dementia in Mice via Metabolic Butyrate. Biomed Res. Int. 2015, 2015, 412946. [Google Scholar] [CrossRef]
- Siracusa, R.; Impellizzeri, D.; Cordaro, M.; Crupi, R.; Esposito, E.; Petrosino, S.; Cuzzocrea, S. Anti-Inflammatory and Neuroprotective Effects of Co-UltraPEALut in a Mouse Model of Vascular Dementia. Front. Neurol. 2017, 8, 233. [Google Scholar] [CrossRef]
- Cai, H.; Cai, T.; Zheng, H.; Liu, L.; Zhou, L.; Pang, X.; Zhan, Q.; Wang, Y.; Yang, C.; Guo, Z.; et al. The Neuroprotective Effects of Danggui-Shaoyao San on Vascular Cognitive Impairment: Involvement of the Role of the Low-Density Lipoprotein Receptor-Related Protein. Rejuvenation Res. 2020, 23, 420–433. [Google Scholar] [CrossRef]
- Zhang, P.; He, S.; Wu, S.; Li, Y.; Wang, H.; Yan, C.; Yang, H.; Li, P. Discovering a Multi-Component Combination Against Vascular Dementia from Danshen-Honghua Herbal Pair by Spectrum-Effect Relationship Analysis. Pharmaceuticals 2022, 15, 1073. [Google Scholar] [CrossRef]
- Hou, X.; Xu, H.; Chen, W.; Zhang, N.; Zhao, Z.; Fang, X.; Zhang, X.; Chen, H.; Xu, Y. Neuroprotective effect of dimethyl fumarate on cognitive impairment induced by ischemic stroke. Ann. Transl. Med. 2020, 8, 375. [Google Scholar] [CrossRef]
- Park, J.A.; Lee, C.H. Neuroprotective Effect of Duloxetine on Chronic Cerebral Hypoperfusion-Induced Hippocampal Neuronal Damage. Biomol. Ther. 2018, 26, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xiao, Y.; Lv, P.; Teng, Z.; Dong, Y.; Qi, Q.; Liu, Z. Edaravone attenuates oxidative stress induced by chronic cerebral hypoperfusion injury: Role of ERK/Nrf2/HO-1 signaling pathway. Neurol. Res. 2018, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, F.; Li, W.; Qin, L.; Yao, Y.; Ge, X.; Yu, Q.; Liang, X.; Zhao, D.; Li, X.; et al. Edaravone injection reverses learning and memory deficits in a rat model of vascular dementia. Acta Biochim. Biophys. Sin. 2017, 49, 83–89. [Google Scholar] [CrossRef]
- Ismaeil, R.; Binti Ahmad Affandi, K.; Kien Hui, C.; Mohamed, W.; Fadly, M. P-BN004. Neuroprotective effect of edible bird’s nest on chronic cerebral hypoperfusion induced neurodegeneration in rats. Clin. Neurophysiol. 2021, 132, e121–e122. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, L.; Li, N.; Dai, C.; Yin, N.; Chu, Z.; Duan, X.; Niu, X.; Yan, P.; Lv, P. Estrogen Exerts Neuroprotective Effects in Vascular Dementia Rats by Suppressing Autophagy and Activating the Wnt/β-Catenin Signaling Pathway. Neurochem. Res. 2020, 45, 2100–2112. [Google Scholar] [CrossRef]
- Kumar, R.; Paracha, V. Neuroprotective effect of genistein on cognitive impairment in vascular dementia experimental mice model. J. Neurol. Sci. 2019, 405, 44. [Google Scholar] [CrossRef]
- Pang, Q.-Q.; Zang, C.-X.; Li, T.; Zeng, X.-C.; Liu, L.-X.; Zhang, D.; Yao, X.-S.; Yu, Y. Neuroprotective effect of GJ-4 against cognitive impairments in vascular dementia by improving white matter damage. Phytomedicine 2024, 132, 155877. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Yang, S.; Song, D.; Wang, C.; Chen, C.; Li, X.; Wang, Q.; Ges, S.; Yang, R.; et al. Ling-Yang-Gou-Teng-decoction prevents vascular dementia through inhibiting oxidative stress induced neurovascular coupling dysfunction. J. Ethnopharmacol. 2018, 222, 229–238. [Google Scholar] [CrossRef]
- Bin-Jaliah, I.; Sakr, H.F. Melatonin ameliorates brain oxidative stress and upregulates senescence marker protein-30 and osteopontin in a rat model of vascular dementia. Physiol. Int. 2018, 105, 38–52. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Gong, Q.; Shi, J.; Li, F. Osthole Improves Cognitive Function of Vascular Dementia Rats: Reducing Aβ Deposition via Inhibition NLRP3 Inflammasome. Biol. Pharm. Bull. 2020, 43, 1315–1323. [Google Scholar] [CrossRef]
- Luo, X.-Q.; Li, A.; Yang, X.; Xiao, X.; Hu, R.; Wang, T.-W.; Dou, X.-Y.; Yang, D.-J.; Dong, Z. Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin. Med. 2018, 13, 14. [Google Scholar] [CrossRef]
- Sun, Z.K.; Ma, X.R.; Jia, Y.J.; Liu, Y.R.; Zhang, J.W.; Zhang, B.A. Effects of resveratrol on apoptosis in a rat model of vascular dementia. Exp. Ther. Med. 2014, 7, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Sunkaria, A.; Singhal, N.; Sandhir, R. Resveratrol loaded solid lipid nanoparticles attenuate mitochondrial oxidative stress in vascular dementia by activating Nrf2/HO-1 pathway. Neurochem. Int. 2018, 112, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.L.; Wu, Z.Z.; Chen, L.X.; Wu, B.X.; Chen, L.L.; Qin, G.C.; Gui, B.; Zhou, J.Y. Neuroprotective effects of tetrandrine against vascular dementia. Neural Regen. Res. 2016, 11, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Sharma, B. Neuroprotective effect of selective DPP-4 inhibitor in experimental vascular dementia. Physiol. Behav. 2015, 152, 182–193. [Google Scholar] [CrossRef]
- Fayez, A.M.; Elnoby, A.S.; Bahnasawy, N.H.; Hassan, O. Neuroprotective effects of zafirlukast, piracetam and their combination on L-Methionine-induced vascular dementia in rats. Fundam. Clin. Pharmacol. 2019, 33, 634–648. [Google Scholar] [CrossRef]
- Jiang, W.; Gong, L.; Liu, F.; Mu, J. Stem cells and vascular dementia: From basic science to the clinic. Cell Tissue Bank. 2020, 21, 349–360. [Google Scholar] [CrossRef]
- He, Y.; Jin, X.; Wang, J.; Meng, M.; Hou, Z.; Tian, W.; Li, Y.; Wang, W.; Wei, Y.; Wang, Y.; et al. Umbilical cord-derived mesenchymal stem cell transplantation for treating elderly vascular dementia. Cell Tissue Bank. 2017, 18, 53–59. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altahrawi, A.Y.; James, A.W.; Shah, Z.A. The Role of Oxidative Stress and Inflammation in the Pathogenesis and Treatment of Vascular Dementia. Cells 2025, 14, 609. https://doi.org/10.3390/cells14080609
Altahrawi AY, James AW, Shah ZA. The Role of Oxidative Stress and Inflammation in the Pathogenesis and Treatment of Vascular Dementia. Cells. 2025; 14(8):609. https://doi.org/10.3390/cells14080609
Chicago/Turabian StyleAltahrawi, Aseel Y., Antonisamy William James, and Zahoor A. Shah. 2025. "The Role of Oxidative Stress and Inflammation in the Pathogenesis and Treatment of Vascular Dementia" Cells 14, no. 8: 609. https://doi.org/10.3390/cells14080609
APA StyleAltahrawi, A. Y., James, A. W., & Shah, Z. A. (2025). The Role of Oxidative Stress and Inflammation in the Pathogenesis and Treatment of Vascular Dementia. Cells, 14(8), 609. https://doi.org/10.3390/cells14080609