
Inflammation-Associated Carcinogenesis in Inflammatory Bowel Disease: Clinical Features and Molecular Mechanisms

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology of Inflammation-Associated Neoplasia in Inflammatory Bowel Disease (IBD)
2.1. UC-Associated Colorectal Neoplasia (UCAN)
2.2. Crohn’s Disease (CD)-Associated Gastrointestinal Neoplasms
3. Risk Factors for Inflammation-Associated Carcinogenesis in IBD
3.1. Patient-Related Risk Factors
3.2. Treatment-Related Risk Factors
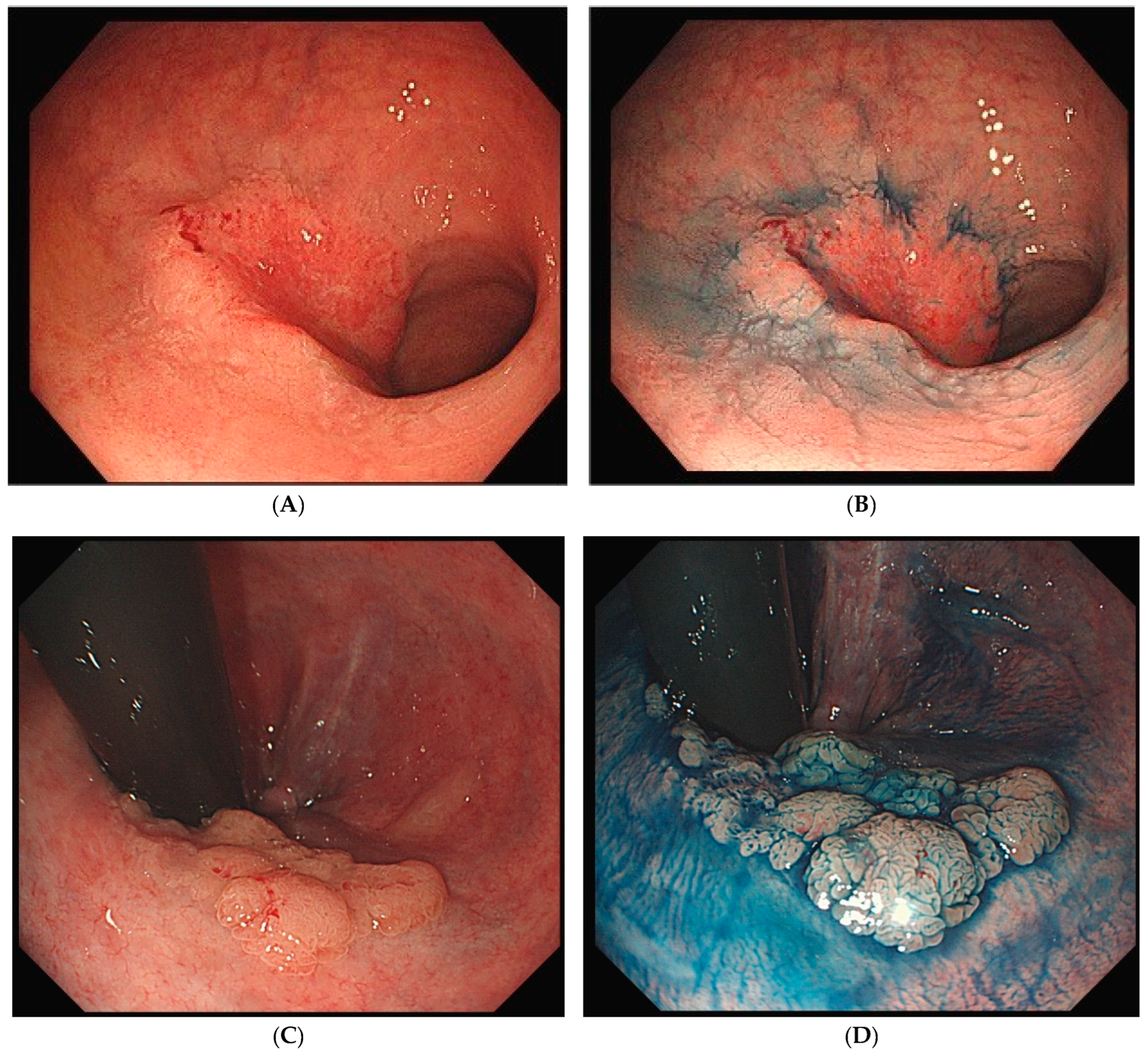
4. Endoscopic and Pathological Features of UCAN
4.1. Endoscopic Features of UCAN
4.1.1. Diagnosis Using White-Light Endoscopy and Chromoendoscopy
4.1.2. Diagnosis Using Magnifying Endoscopy and IEE
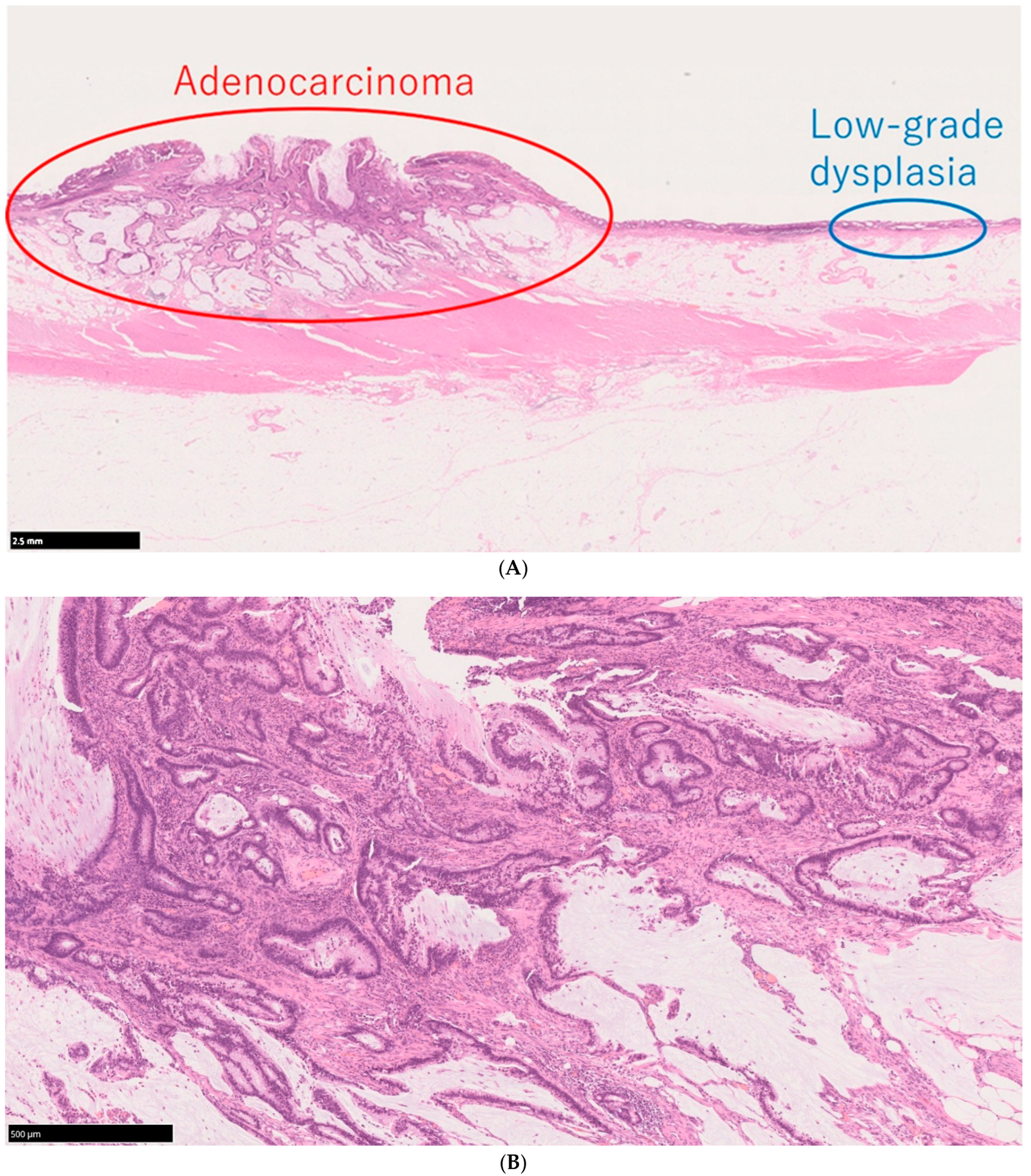
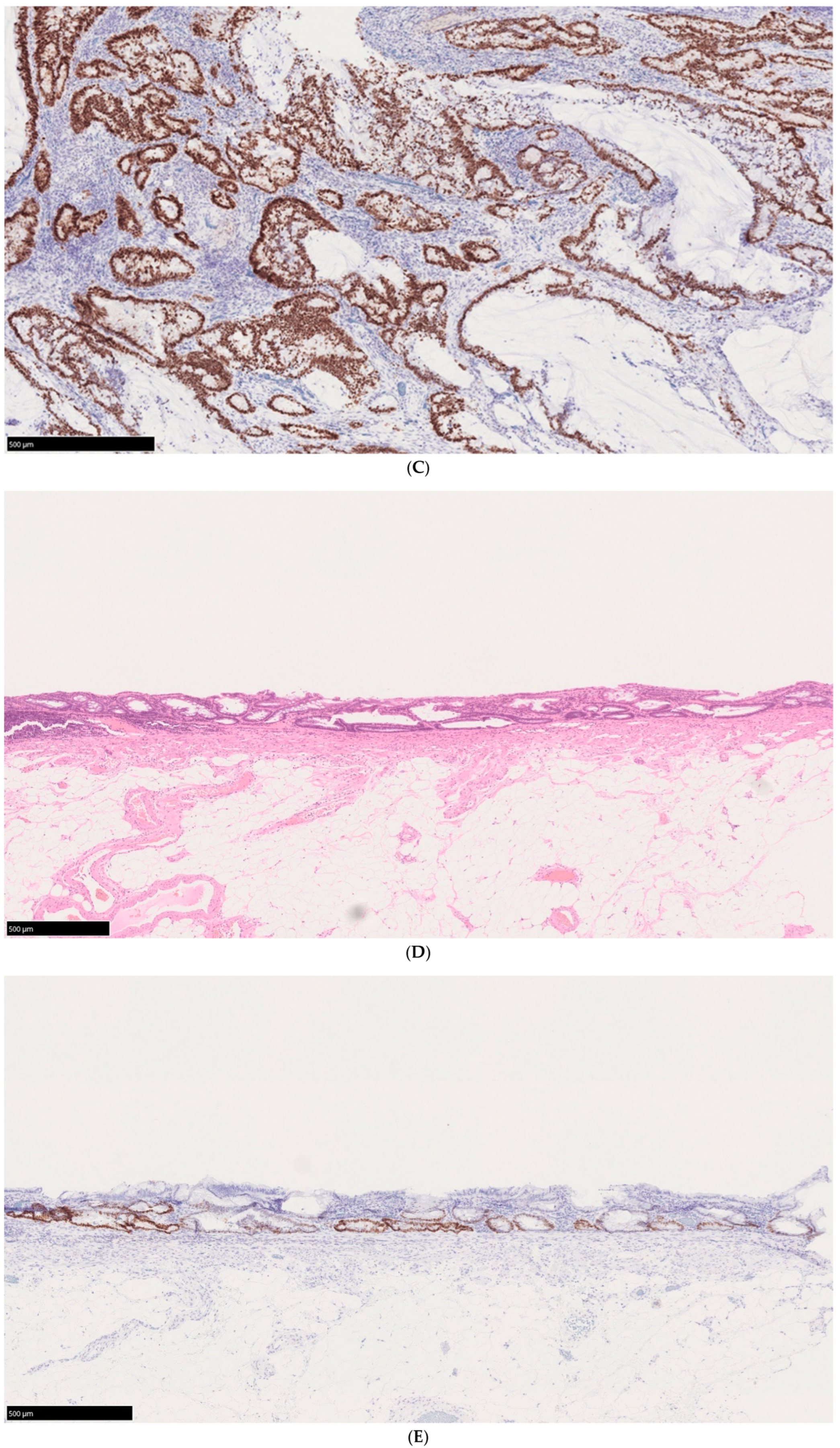
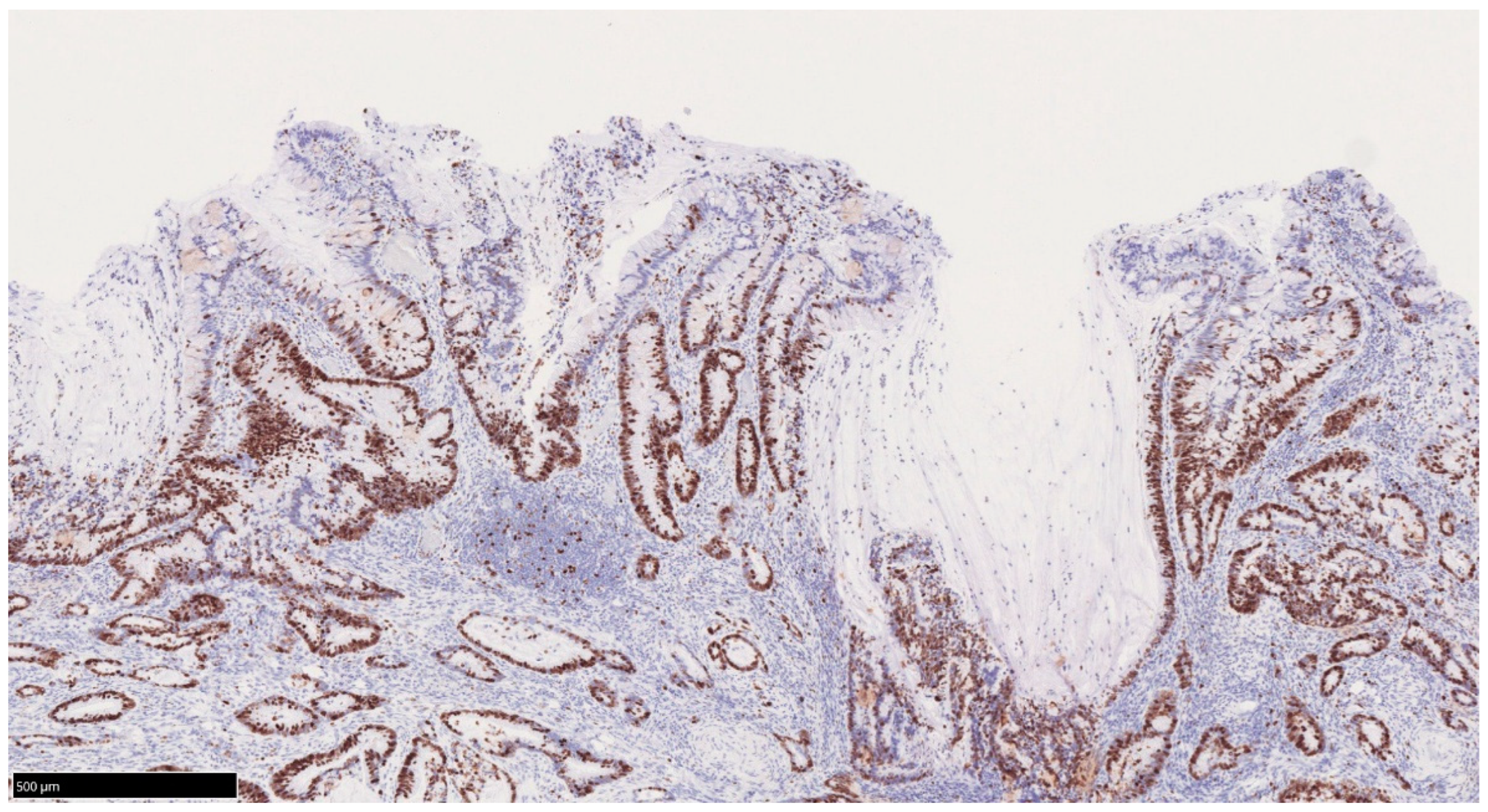
4.2. Pathological Features of UCAN
5. Surveillance Colonoscopy: Current Status and Challenges
5.1. Targets for Surveillance Colonoscopy
5.2. Timing and Methods of Surveillance Colonoscopy
5.3. IEE for Detection of UCAN
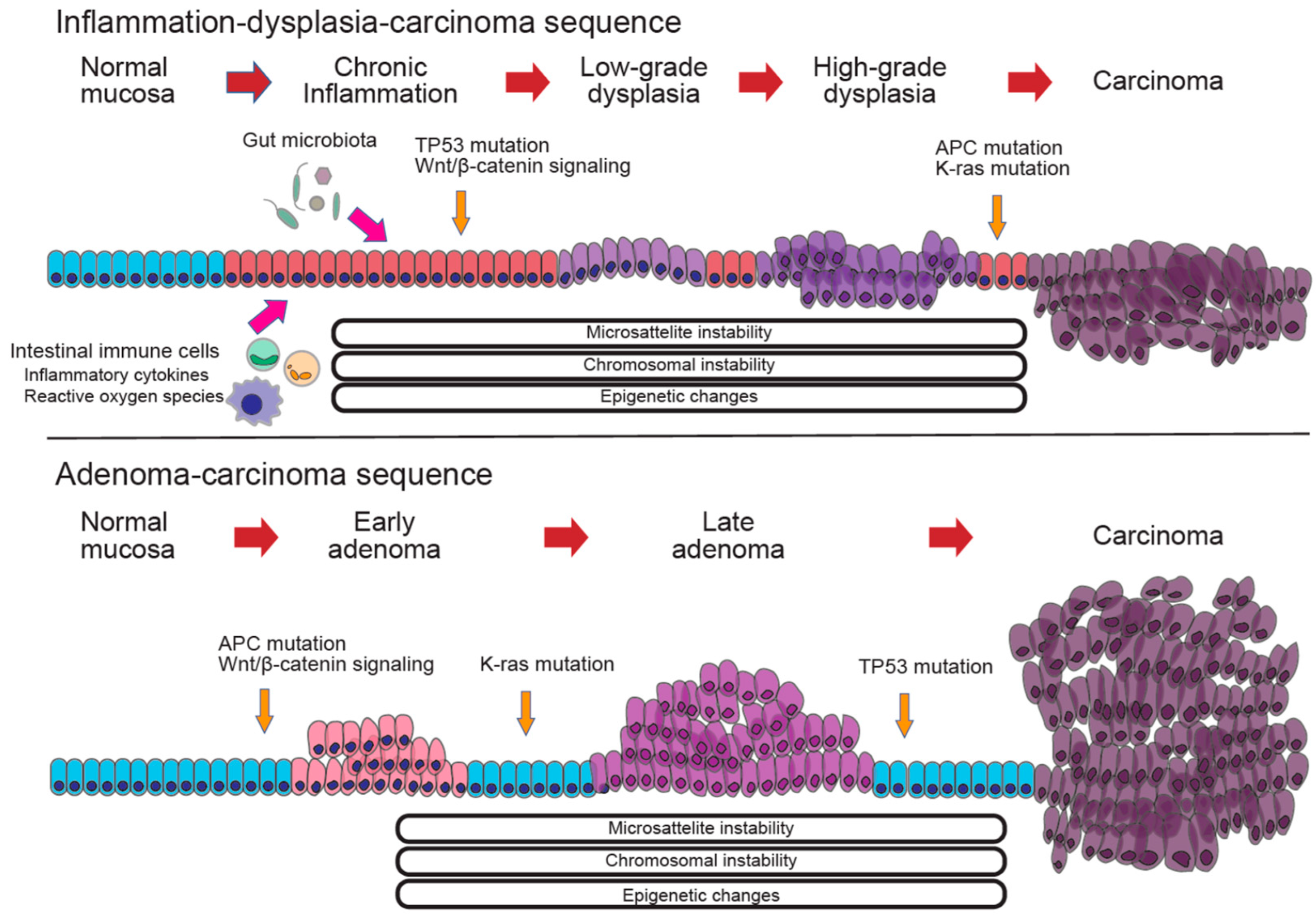
6. Molecular Mechanisms of Colitis-Associated Cancer (CAC) in IBD
6.1. Molecular Pathways in CAC
6.2. Genetic Alterations and Epigenetic Changes in CAC
6.3. Genetic Drivers of CAC
6.4. The Role of Gut Microbiota in CAC
6.5. Animal Models for CAC Research
6.6. Organoid Models for CAC Research
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turner, D.; Ricciuto, A.; Lewis, A.; D’Amico, F.; Dhaliwal, J.; Griffiths, A.M.; Bettenworth, D.; Sandborn, W.J.; Sands, B.E.; Reinisch, W.; et al. International Organization for the Study of IBD. STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology 2021, 160, 1570–1583. [Google Scholar] [PubMed]
- Tsai, L.; Ma, C.; Dulai, P.S.; Prokop, L.J.; Eisenstein, S.; Ramamoorthy, S.L.; Feagan, B.G.; Jairath, V.; Sandborn, W.J.; Singh, S. Contemporary Risk of Surgery in Patients with Ulcerative Colitis and Crohn’s Disease: A Meta-Analysis of Population-Based Cohorts. Clin. Gastroenterol. Hepatol. 2021, 19, 2031–2045.e11. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Rutter, M.D.; Askari, A.; Lee, G.H.; Warusavitarne, J.; Moorghen, M.; Thomas-Gibson, S.; Saunders, B.P.; Graham, T.A.; Hart, A.L. Forty-Year Analysis of Colonoscopic Surveillance Program for Neoplasia in Ulcerative Colitis: An Updated Overview. Am. J. Gastroenterol. 2015, 110, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, J.; Hata, K.; Kazama, S.; Anzai, H.; Shinagawa, T.; Murono, K.; Kaneko, M.; Sasaki, K.; Yasuda, K.; Otani, K.; et al. Results of a 36-year surveillance program for ulcerative colitis-associated neoplasia in the Japanese population. Dig. Endosc. 2018, 30, 236–244. [Google Scholar] [CrossRef]
- Lutgens, M.W.; van Oijen, M.G.; van der Heijden, G.J.; Vleggaar, F.P.; Siersema, P.D.; Oldenburg, B. Declining risk of colorectal cancer in inflammatory bowel disease: An updated meta-analysis of population-based cohort studies. Inflamm. Bowel Dis. 2013, 19, 789–799. [Google Scholar] [CrossRef]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal cancer in ulcerative colitis: A Scandinavian population-based cohort study. Lancet 2020, 395, 123–131. [Google Scholar] [CrossRef]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef]
- Jess, T.; Gamborg, M.; Matzen, P.; Munkholm, P.; Sorensen, T.I. Increased risk of intestinal cancer in Crohn’s disease: A meta-analysis of population-based cohort studies. Am. J. Gastroenterol. 2005, 100, 2724–2729. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Piovani, D.; Hassan, C.; Repici, A.; Rimassa, L.; Carlo-Stella, C.; Nikolopoulos, G.K.; Riboli, E.; Bonovas, S. Risk of Cancer in Inflammatory Bowel Diseases: Umbrella Review and Reanalysis of Meta-analyses. Gastroenterology 2022, 163, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Zaanan, A.; Svrcek, M.; Laurent-Puig, P.; Carrere, N.; Manfredi, S.; Locher, C.; Afchain, P. Small bowel adenocarcinoma: Epidemiology, risk factors, diagnosis and treatment. Dig. Liver Dis. 2014, 46, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Laukoetter, M.G.; Mennigen, R.; Hannig, C.M.; Osada, N.; Rijcken, E.; Vowinkel, T.; Krieglstein, C.F.; Senninger, N.; Anthoni, C.; Bruewer, M. Intestinal cancer risk in Crohn’s disease: A meta-analysis. J. Gastrointest. Surg. 2011, 15, 576–583. [Google Scholar] [CrossRef]
- Gordon, H.; Biancone, L.; Fiorino, G.; Katsanos, K.H.; Kopylov, U.; Al Sulais, E.; E Axelrad, J.; Balendran, K.; Burisch, J.; de Ridder, L.; et al. ECCO Guidelines on Inflammatory Bowel Disease and Malignancies. J. Crohn’s Colitis 2023, 17, 827–854. [Google Scholar] [CrossRef]
- Choi, P.M.; Zelig, M.P. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: Implications for carcinogenesis and prevention. Gut 1994, 35, 950–954. [Google Scholar] [CrossRef]
- Delaunoit, T.; Limburg, P.J.; Goldberg, R.M.; Lymp, J.F.; Loftus, E.V. Colorectal cancer prognosis among patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2006, 4, 335–342. [Google Scholar] [CrossRef]
- Noguchi, T.; Ishihara, S.; Uchino, M.; Ikeuchi, H.; Okabayashi, K.; Futami, K.; Tanaka, S.; Ohge, H.; Nagahara, H.; Watanabe, K.; et al. Clinical features and oncological outcomes of intestinal cancers associated with ulcerative colitis and Crohn’s disease. J. Gastroenterol. 2023, 58, 14–24. [Google Scholar] [CrossRef]
- Uchino, M.; Ikeuchi, H.; Hata, K.; Minagawa, T.; Horio, Y.; Kuwahara, R.; Nakamura, S.; Watanabe, K.; Saruta, M.; Fujii, T.; et al. Intestinal cancer in patients with Crohn’s disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2021, 36, 329–336. [Google Scholar] [CrossRef]
- van Rheenen, P.F.; Aloi, M.; Biron, I.A.; Carlsen, K.; Cooney, R.; Cucchiara, S.; Cullen, G.; Escher, J.C.; Kierkus, J.; Lindsay, J.O.; et al. European Crohn’s and Colitis Organisation Topical Review on Transitional Care in Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 1032–1038. [Google Scholar] [CrossRef]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, A.M.; de Jong, M.E.; Lutgens, M.; Hoentjen, F.; Elias, S.G.; Oldenburg, B. Prognostic Factors for Advanced Colorectal Neoplasia in Inflammatory Bowel Disease: Systematic Review and Meta-analysis. Gastroenterology 2021, 160, 1584–1598. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Dickman, P.W.; Karlén, P.; Broström, O.; Lapidus, A.; Löfberg, R.; Ekbom, A. Colorectal cancer rates among first-degree relatives of patients with inflammatory bowel disease: A population-based cohort study. Lancet 2001, 357, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Farraye, F.A.; Odze, R.D.; Eaden, J.; Itzkowitz, S.H. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology 2010, 138, 746–774, 774.e1–4, quiz e12–13. [Google Scholar] [CrossRef]
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e713. [Google Scholar] [CrossRef]
- Shah, S.C.; Ten Hove, J.R.; Castaneda, D.; Palmela, C.; Mooiweer, E.; Colombel, J.F.; Harpaz, N.; Ullman, T.A.; van Bodegraven, A.A.; Jansen, J.M.; et al. High Risk of Advanced Colorectal Neoplasia in Patients With Primary Sclerosing Cholangitis Associated With Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 1106–1113.e1103. [Google Scholar] [CrossRef]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: A population-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
- Axelrad, J.E.; Olén, O.; Sachs, M.C.; Erichsen, R.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Inflammatory bowel disease and risk of small bowel cancer: A binational population-based cohort study from Denmark and Sweden. Gut 2021, 70, 297–308. [Google Scholar]
- Hata, K.; Anzai, H.; Ikeuchi, H.; Futami, K.; Fukushima, K.; Sugita, A.; Uchino, M.; Higashi, D.; Itabashi, M.; Watanabe, K.; et al. Surveillance Colonoscopy for Ulcerative Colitis-Associated Colorectal Cancer Offers Better Overall Survival in Real-World Surgically Resected Cases. Am. J. Gastroenterol. 2019, 114, 483–489. [Google Scholar] [CrossRef]
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Cancer surveillance in longstanding ulcerative colitis: Endoscopic appearances help predict cancer risk. Gut 2004, 53, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, M.M.; Chen, M.; Theeuwes, M.; van Dekken, H.; Sikkema, M.; Steyerberg, E.W.; Lingsma, H.F.; Siersema, P.D.; Xia, B.; Kusters, J.G.; et al. Biomarker-based prediction of inflammatory bowel disease-related colorectal cancer: A case-control study. Cell. Oncol. 2011, 34, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Rahier, J.F.; Kirchgesner, J. Predicting, Preventing, and Managing Treatment-Related Complications in Patients With Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 1324–1335.e1322. [Google Scholar] [CrossRef]
- Velayos, F.S.; Terdiman, J.P.; Walsh, J.M. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. Am. J. Gastroenterol. 2005, 100, 1345–1353. [Google Scholar] [CrossRef]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019, 68 (Suppl. S3), s1–s106. [Google Scholar] [CrossRef]
- Laine, L.; Kaltenbach, T.; Barkun, A.; McQuaid, K.R.; Subramanian, V.; Soetikno, R.; SCENIC Guideline Development Panel. SCENIC international consensus statement on surveillance and management of dysplasia in inflammatory bowel disease. Gastrointest. Endosc. 2015, 81, 489–501. [Google Scholar] [CrossRef]
- Sugimoto, S.; Naganuma, M.; Iwao, Y.; Matsuoka, K.; Shimoda, M.; Mikami, S.; Mizuno, S.; Nakazato, Y.; Nanki, K.; Inoue, N.; et al. Endoscopic morphologic features of ulcerative colitis-associated dysplasia classified according to the SCENIC consensus statement. Gastrointest. Endosc. 2017, 85, 639–646. [Google Scholar] [CrossRef]
- Matsumoto, T.; Iwao, Y.; Igarashi, M.; Watanabe, K.; Otsuka, K.; Watanabe, T.; Iizuka, B.; Hida, N.; Sada, M.; Chiba, T.; et al. Endoscopic and chromoendoscopic atlas featuring dysplastic lesions in surveillance colonoscopy for patients with long-standing ulcerative colitis. Inflamm. Bowel Dis. 2008, 14, 259–264. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Ohno, A.; Chiba, T. Linked color imaging identified ulcerative colitis-associated colorectal cancer: A case report. Dig. Endosc. 2018, 30, 267. [Google Scholar] [CrossRef]
- Nishiyama, S.; Oka, S.; Tanaka, S.; Sagami, S.; Hayashi, R.; Ueno, Y.; Arihiro, K.; Chayama, K. Clinical usefulness of narrow band imaging magnifying colonoscopy for assessing ulcerative colitis-associated cancer/dysplasia. Endosc. Int. Open 2016, 4, E1183–E1187. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Tanaka, S.; Kudo, S.; Saito, S.; Matsuda, T.; Wada, Y.; Fujii, T.; Ikematsu, H.; Uraoka, T.; Kobayashi, N.; et al. Narrow-band imaging (NBI) magnifying endoscopic classification of colorectal tumors proposed by the Japan NBI Expert Team. Dig. Endosc. 2016, 28, 526–533. [Google Scholar] [CrossRef]
- Kawasaki, K.; Nakamura, S.; Esaki, M.; Kurahara, K.; Eizuka, M.; Nuki, Y.; Kochi, S.; Fujiwara, M.; Oshiro, Y.; Sugai, T.; et al. Clinical usefulness of magnifying colonoscopy for the diagnosis of ulcerative colitis-associated neoplasia. Dig. Endosc. 2019, 31 (Suppl. S1), 36–42. [Google Scholar] [CrossRef]
- Kudo, S.; Maeda, Y.; Ogata, N.; Misawa, M.; Ogawa, Y.; Takishima, K.; Ishiyama, M.; Mochizuki, K.; Minegishi, Y.; Ogura, Y.; et al. Combined endocytoscopy with pit pattern diagnosis in ulcerative colitis-associated neoplasia: Pilot study. Dig. Endosc. 2022, 34, 133–143. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumors Ediotorial Board. Digestive System Tumours, WHO Classification of Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1. [Google Scholar]
- Lu, X.; Yu, Y.; Tan, S. p53 expression in patients with ulcerative colitis–associated with dysplasia and carcinoma: A systematic meta-analysis. BMC Gastroenterol. 2017, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.N.; Rognum, T.O.; Bakka, A.; Clausen, O.P. Ki-67: A useful marker for the evaluation of dysplasia in ulcerative colitis. Mol. Pathol. 1998, 51, 327–332. [Google Scholar] [CrossRef]
- Riddell, R.H.; Goldman, H.; Ransohoff, D.F.; Appelman, H.D.; Fenoglio, C.M.; Haggitt, R.C.; Ahren, C.; Correa, P.; Hamilton, S.R.; Morson, B.C.; et al. Dysplasia in inflammatory bowel disease: Standardized classification with provisional clinical applications. Hum. Pathol. 1983, 14, 931–968. [Google Scholar] [CrossRef]
- Sugimoto, S.; Shimoda, M.; Iwao, Y.; Mutaguchi, M.; Nanki, K.; Mizuno, S.; Kameyama, K.; Ogata, H.; Naganuma, M.; Kanai, T. Intramucosal poorly differentiated and signet-ring cell components in patients with ulcerative colitis-associated high-grade dysplasia. Dig. Endosc. 2019, 31, 706–711. [Google Scholar] [CrossRef]
- Mutaguchi, M.; Naganuma, M.; Sugimoto, S.; Fukuda, T.; Nanki, K.; Mizuno, S.; Hosoe, N.; Shimoda, M.; Ogata, H.; Iwao, Y.; et al. Difference in the clinical characteristic and prognosis of colitis-associated cancer and sporadic neoplasia in ulcerative colitis patients. Dig. Liver Dis. 2019, 51, 1257–1264. [Google Scholar] [CrossRef]
- Matsuoka, K.; Kobayashi, T.; Ueno, F.; Matsui, T.; Hirai, F.; Inoue, N.; Kato, J.; Kobayashi, K.; Kobayashi, K.; Koganei, K.; et al. Evidence-based clinical practice guidelines for inflammatory bowel disease. J. Gastroenterol. 2018, 53, 305–353. [Google Scholar] [CrossRef]
- Watanabe, T.; Ajioka, Y.; Mitsuyama, K.; Watanabe, K.; Hanai, H.; Nakase, H.; Kunisaki, R.; Matsuda, K.; Iwakiri, R.; Hida, N.; et al. Comparison of Targeted vs Random Biopsies for Surveillance of Ulcerative Colitis-Associated Colorectal Cancer. Gastroenterology 2016, 151, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Moussata, D.; Allez, M.; Cazals-Hatem, D.; Treton, X.; Laharie, D.; Reimund, J.M.; Bertheau, P.; Bourreille, A.; Lavergne-Slove, A.; Brixi, H.; et al. Are random biopsies still useful for the detection of neoplasia in patients with IBD undergoing surveillance colonoscopy with chromoendoscopy? Gut 2018, 67, 616–624. [Google Scholar] [CrossRef]
- Murthy, S.K.; Feuerstein, J.D.; Nguyen, G.C.; Velayos, F.S. AGA Clinical Practice Update on Endoscopic Surveillance and Management of Colorectal Dysplasia in Inflammatory Bowel Diseases: Expert Review. Gastroenterology 2021, 161, 1043–1051.e4. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology 2006, 130, 1030–1038. [Google Scholar] [CrossRef]
- Har-Noy, O.; Katz, L.; Avni, T.; Battat, R.; Bessissow, T.; Yung, D.E.; Engel, T.; Koulaouzidis, A.; Eliakim, R.; Ben-Horin, S.; et al. Chromoendoscopy, Narrow-Band Imaging or White Light Endoscopy for Neoplasia Detection in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2017, 62, 2982–2990. [Google Scholar] [CrossRef]
- Alexandersson, B.; Hamad, Y.; Andreasson, A.; Rubio, C.A.; Ando, Y.; Tanaka, K.; Ichiya, T.; Rezaie, R.; Schmidt, P.T. High-Definition Chromoendoscopy Superior to High-Definition White-Light Endoscopy in Surveillance of Inflammatory Bowel Diseases in a Randomized Trial. Clin. Gastroenterol. Hepatol. 2020, 18, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.H.; Park, S.J.; Kim, H.S.; Park, Y.S.; Park, D.I.; Lee, K.M.; Jung, S.A.; Choi, C.H.; Koo, J.S.; Cheon, J.H.; et al. High-Definition Chromoendoscopy Versus High-Definition White Light Colonoscopy for Neoplasia Surveillance in Ulcerative Colitis: A Randomized Controlled Trial. Am. J. Gastroenterol. 2019, 114, 1642–1648. [Google Scholar] [CrossRef]
- Feuerstein, J.D.; Rakowsky, S.; Sattler, L.; Yadav, A.; Foromera, J.; Grossberg, L.; Cheifetz, A.S. Meta-analysis of dye-based chromoendoscopy compared with standard- and high-definition white-light endoscopy in patients with inflammatory bowel disease at increased risk of colon cancer. Gastrointest. Endosc. 2019, 90, 186–195.e1. [Google Scholar] [CrossRef]
- Resende, R.H.; Ribeiro, I.B.; de Moura, D.T.H.; Galetti, F.; Rocha, R.S.P.; Bernardo, W.M.; Sakai, P.; de Moura, E.G.H. Surveillance in inflammatory bowel disease: Is chromoendoscopy the only way to go? A systematic review and meta-analysis of randomized clinical trials. Endosc. Int. Open 2020, 8, E578–E590. [Google Scholar] [CrossRef]
- Gondal, B.; Haider, H.; Komaki, Y.; Komaki, F.; Micic, D.; Rubin, D.T.; Sakuraba, A. Efficacy of various endoscopic modalities in detecting dysplasia in ulcerative colitis: A systematic review and network meta-analysis. World J. Gastrointest. Endosc. 2020, 12, 159–171. [Google Scholar] [CrossRef]
- El-Dallal, M.; Chen, Y.; Lin, Q.; Rakowsky, S.; Sattler, L.; Foromera, J.; Grossberg, L.; Cheifetz, A.S.; Feuerstein, J.D. Meta-analysis of Virtual-based Chromoendoscopy Compared With Dye-spraying Chromoendoscopy Standard and High-definition White Light Endoscopy in Patients With Inflammatory Bowel Disease at Increased Risk of Colon Cancer. Inflamm. Bowel Dis. 2020, 26, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Sato, T.; Kamikoduru, K. Surveillance colonoscopy with pancolonic narrow band imaging observation in patients with ulcerative colitis. Gastroenterol. Endosc. 2020, 62, 2972–2979. [Google Scholar]
- Takabayashi, K.; Kato, M.; Sugimoto, S.; Yahagi, N.; Kanai, T. Texture and color enhancement imaging in combination with indigo carmine dye spraying to highlight the border of flat ulcerative colitis-associated neoplasia. Gastrointest. Endosc. 2022, 95, 1273–1275. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.M.; Nusrat, A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Hyun, Y.S.; Han, D.S.; Lee, A.R.; Eun, C.S.; Youn, J.; Kim, H.Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nakamura, T.; Takenouchi, S.; Hayashi, A.; Omori, K.; Murata, T. Urinary 8-iso PGF2α and 2,3-dinor-8-iso PGF2α can be indexes of colitis-associated colorectal cancer in mice. PLoS ONE 2021, 16, e0245292. [Google Scholar] [CrossRef]
- Garg, P.; Sarma, D.; Jeppsson, S.; Patel, N.R.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. Matrix metalloproteinase-9 functions as a tumor suppressor in colitis-associated cancer. Cancer Res. 2010, 70, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Sarkar, S.; Wang, Y.; Singh, P. Functional cross-talk between beta-catenin and NFkappaB signaling pathways in colonic crypts of mice in response to progastrin. J. Biol. Chem. 2009, 284, 22274–22284. [Google Scholar] [CrossRef]
- Oguma, K.; Oshima, H.; Aoki, M.; Uchio, R.; Naka, K.; Nakamura, S.; Hirao, A.; Saya, H.; Taketo, M.M.; Oshima, M. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008, 27, 1671–1681. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tomita, H.; Shimizu, M.; Tanaka, T.; Suzui, N.; Miyazaki, T.; Hara, A. p53 Expression as a Diagnostic Biomarker in Ulcerative Colitis-Associated Cancer. Int. J. Mol. Sci. 2017, 18, 1284. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6. [Google Scholar] [CrossRef]
- Du, L.; Kim, J.J.; Shen, J.; Chen, B.; Dai, N. KRAS and TP53 mutations in inflammatory bowel disease-associated colorectal cancer: A meta-analysis. Oncotarget 2017, 8, 22175–22186. [Google Scholar] [CrossRef]
- Itzkowitz, S.H. Molecular biology of dysplasia and cancer in inflammatory bowel disease. Gastroenterol. Clin. N. Am. 2006, 35, 553–571. [Google Scholar] [CrossRef]
- Rubin, C.E.; Haggitt, R.C.; Burmer, G.C.; Brentnall, T.A.; Stevens, A.C.; Levine, D.S.; Dean, P.J.; Kimmey, M.; Perera, D.R.; Rabinovitch, P.S. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992, 103, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Olafsson, S.; McIntyre, R.E.; Coorens, T.; Butler, T.; Jung, H.; Robinson, P.S.; Lee-Six, H.; Sanders, M.A.; Arestang, K.; Dawson, C.; et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 2020, 182, 672–684.e11. [Google Scholar] [CrossRef]
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Boland, C.R. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, C148–C154. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Inoue, N.; Hisamatsu, T.; Kashiwagi, K.; Takaishi, H.; Kanai, T.; Watanabe, M.; Ishii, H.; Hibi, T. Clinical significance of microsatellite instability in the inflamed mucosa for the prediction of colonic neoplasms in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 2005, 20, 710–715. [Google Scholar] [CrossRef]
- Scarpa, M.; Scarpa, M.; Castagliuolo, I.; Erroi, F.; Kotsafti, A.; Basato, S.; Brun, P.; D’Incà, R.; Rugge, M.; Angriman, I.; et al. Aberrant gene methylation in non-neoplastic mucosa as a predictive marker of ulcerative colitis-associated CRC. Oncotarget 2016, 7, 10322–10331. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Esteller, M.; Harpaz, N.; Leytin, A.; Rashid, A.; Xu, Y.; Liang, J.; Stine, O.C.; Yin, J.; Zou, T.T.; et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000, 60, 4864–4868. [Google Scholar]
- Meyers, M.; Wagner, M.W.; Hwang, H.S.; Kinsella, T.J.; Boothman, D.A. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001, 61, 5193–5201. [Google Scholar] [PubMed]
- Fujii, S.; Tominaga, K.; Kitajima, K.; Takeda, J.; Kusaka, T.; Fujita, M.; Ichikawa, K.; Tomita, S.; Ohkura, Y.; Ono, Y.; et al. Methylation of the oestrogen receptor gene in non-neoplastic epithelium as a marker of colorectal neoplasia risk in longstanding and extensive ulcerative colitis. Gut 2005, 54, 1287–1292. [Google Scholar] [CrossRef]
- Issa, J.P.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Fujita, M.; Matsubara, N.; Matsuda, I.; Maejima, K.; Oosawa, A.; Yamano, T.; Fujimoto, A.; Furuta, M.; Nakano, K.; Oku-Sasaki, A.; et al. Genomic landscape of colitis-associated cancer indicates the impact of chronic inflammation and its stratification by mutations in the Wnt signaling. Oncotarget 2018, 9, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Rajamäki, K.; Taira, A.; Katainen, R.; Välimäki, N.; Kuosmanen, A.; Plaketti, R.M.; Seppälä, T.T.; Ahtiainen, M.; Wirta, E.V.; Vartiainen, E.; et al. Genetic and Epigenetic Characteristics of Inflammatory Bowel Disease-Associated Colorectal Cancer. Gastroenterology 2021, 161, 592–607. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Lee, Y.R.; Lee, A.R.; Park, C.H.; Han, D.S.; Eun, C.S. Role of the global gut microbial community in the development of colitis-associated cancer in a murine model. Biomed. Pharmacother. 2021, 135, 111206. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e324. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Vila, A.V.; Gacesa, R.; Collij, V.; Stevens, C.; Fu, J.M.; Wong, I.; Talkowski, M.E.; Rivas, M.A.; Imhann, F.; et al. Whole exome sequencing analyses reveal gene-microbiota interactions in the context of IBD. Gut 2021, 70, 285–296. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T. The Potential Role of Exercise and Nutrition in Harnessing the Immune System to Improve Colorectal Cancer Survival. Gastroenterology 2018, 155, 596–600. [Google Scholar] [CrossRef]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Asp. Med. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Modesto, R.; Estarreja, J.; Silva, I.; Rocha, J.; Pinto, R.; Mateus, V. Induced Colitis-Associated Cancer Models in Rodents for Pharmacological Modulation: A Systematic Review. J. Clin. Med. 2022, 11, 2739. [Google Scholar] [CrossRef]
- Tang, A.; Li, N.; Li, X.; Yang, H.; Wang, W.; Zhang, L.; Li, G.; Xiong, W.; Ma, J.; Shen, S. Dynamic activation of the key pathways: Linking colitis to colorectal cancer in a mouse model. Carcinogenesis 2012, 33, 1375–1383. [Google Scholar] [CrossRef]
- Uragami, T.; Ando, Y.; Aoi, M.; Fukui, T.; Matsumoto, Y.; Horitani, S.; Tomiyama, T.; Okazaki, K.; Tsuneyama, K.; Tanaka, H.; et al. Establishment of a Novel Colitis-Associated Cancer Mouse Model Showing Flat Invasive Neoplasia. Dig. Dis. Sci. 2023, 68, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Fujii, M.; Shimokawa, M.; Matano, M.; Nishikori, S.; Date, S.; Takano, A.; Toshimitsu, K.; Ohta, Y.; Takahashi, S.; et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 2020, 577, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hisamatsu, T.; Miyoshi, J.; Oguri, N.; Morikubo, H.; Saito, D.; Hayashi, A.; Omori, T.; Matsuura, M. Inflammation-Associated Carcinogenesis in Inflammatory Bowel Disease: Clinical Features and Molecular Mechanisms. Cells 2025, 14, 567. https://doi.org/10.3390/cells14080567
Hisamatsu T, Miyoshi J, Oguri N, Morikubo H, Saito D, Hayashi A, Omori T, Matsuura M. Inflammation-Associated Carcinogenesis in Inflammatory Bowel Disease: Clinical Features and Molecular Mechanisms. Cells. 2025; 14(8):567. https://doi.org/10.3390/cells14080567
Chicago/Turabian StyleHisamatsu, Tadakazu, Jun Miyoshi, Noriaki Oguri, Hiromu Morikubo, Daisuke Saito, Akimasa Hayashi, Teppei Omori, and Minoru Matsuura. 2025. "Inflammation-Associated Carcinogenesis in Inflammatory Bowel Disease: Clinical Features and Molecular Mechanisms" Cells 14, no. 8: 567. https://doi.org/10.3390/cells14080567
APA StyleHisamatsu, T., Miyoshi, J., Oguri, N., Morikubo, H., Saito, D., Hayashi, A., Omori, T., & Matsuura, M. (2025). Inflammation-Associated Carcinogenesis in Inflammatory Bowel Disease: Clinical Features and Molecular Mechanisms. Cells, 14(8), 567. https://doi.org/10.3390/cells14080567