
GOLPH3-mTOR Crosstalk and Glycosylation: A Molecular Driver of Cancer Progression



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: GOLPH3, a PI(4)P-Binding Oncoprotein
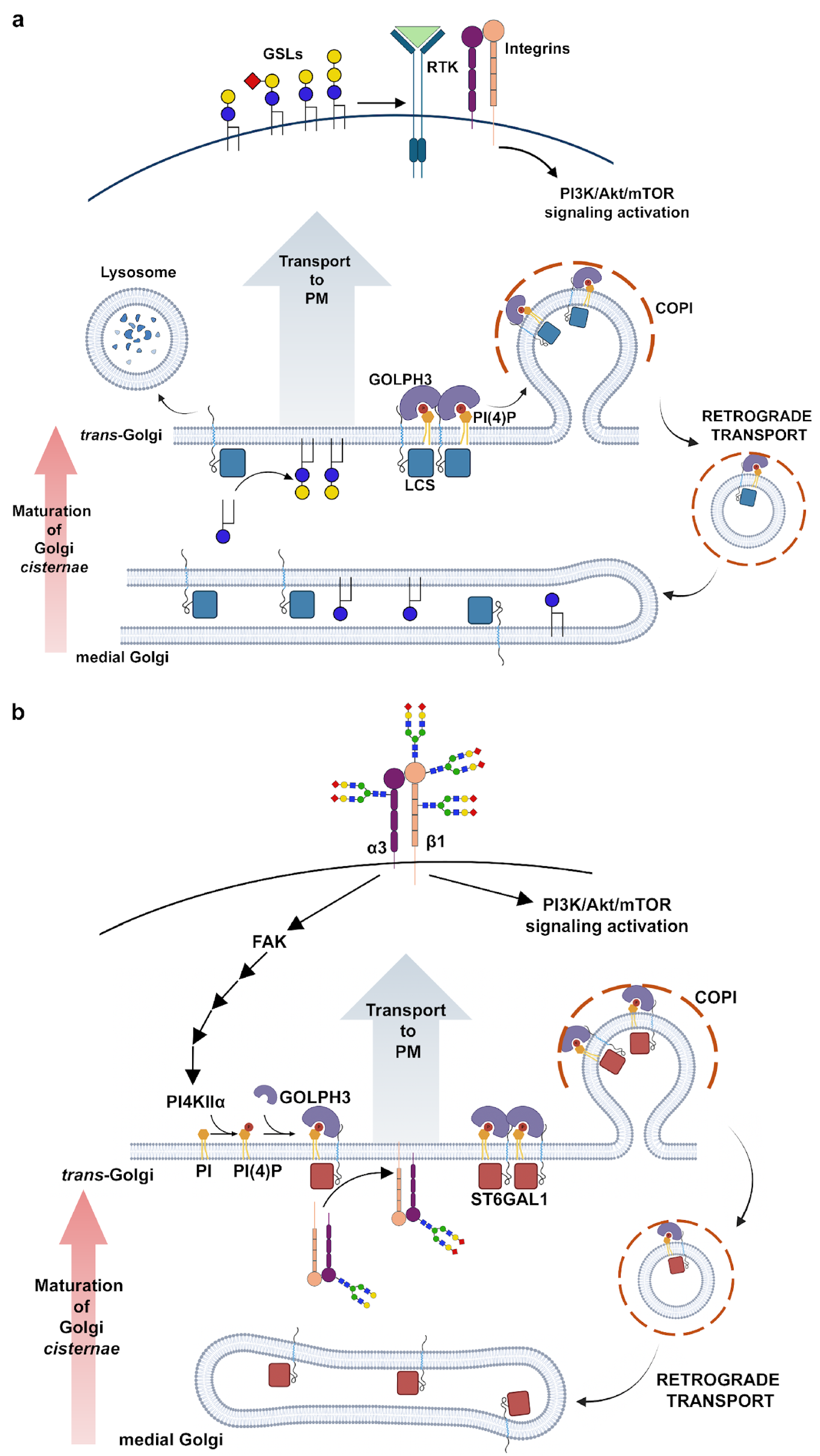
2. Role of GOLPH3 Family Proteins in Golgi Structure Maintenance and Glycosylation
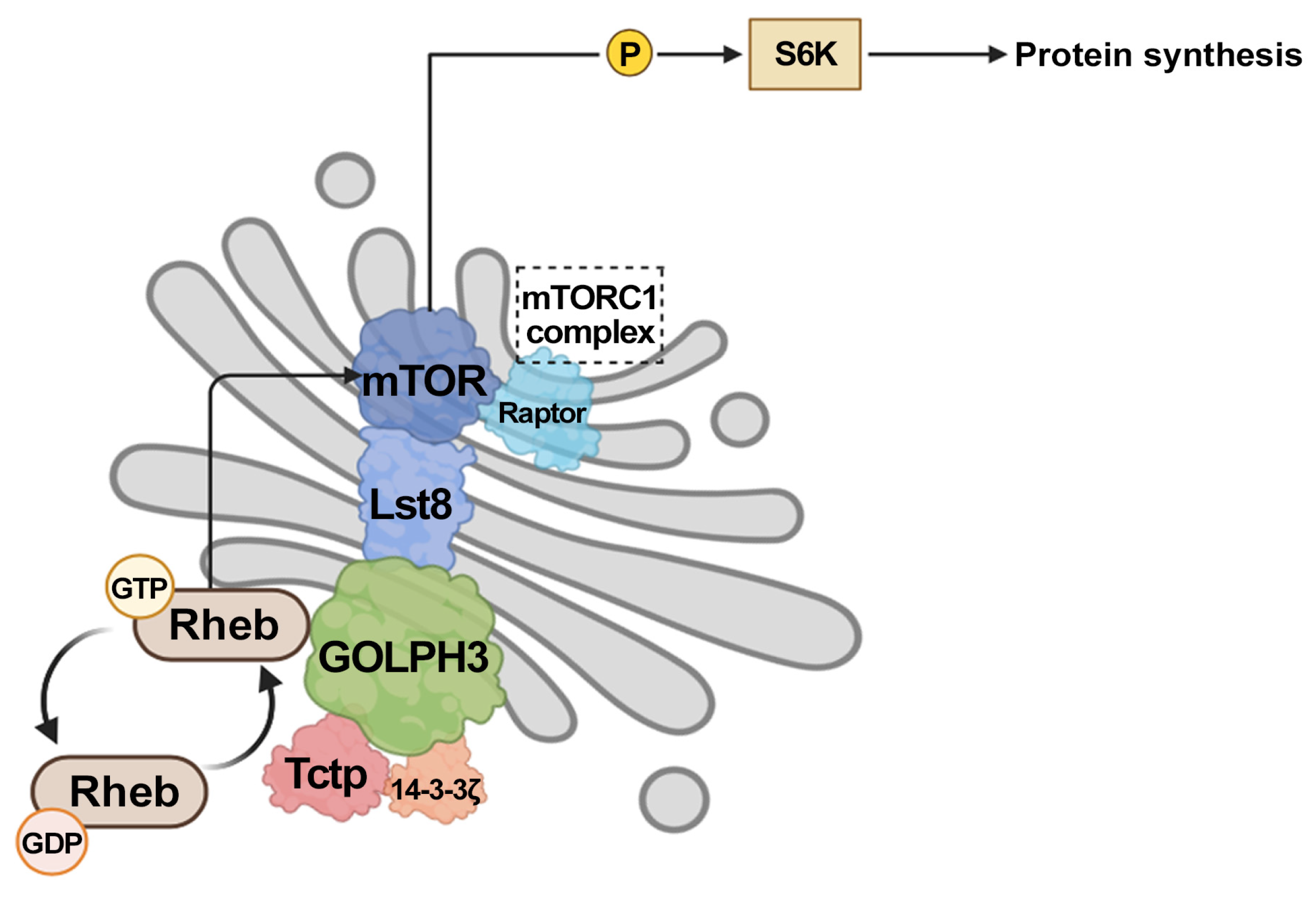
3. GOLPH3 Enhances Signaling Through mTOR
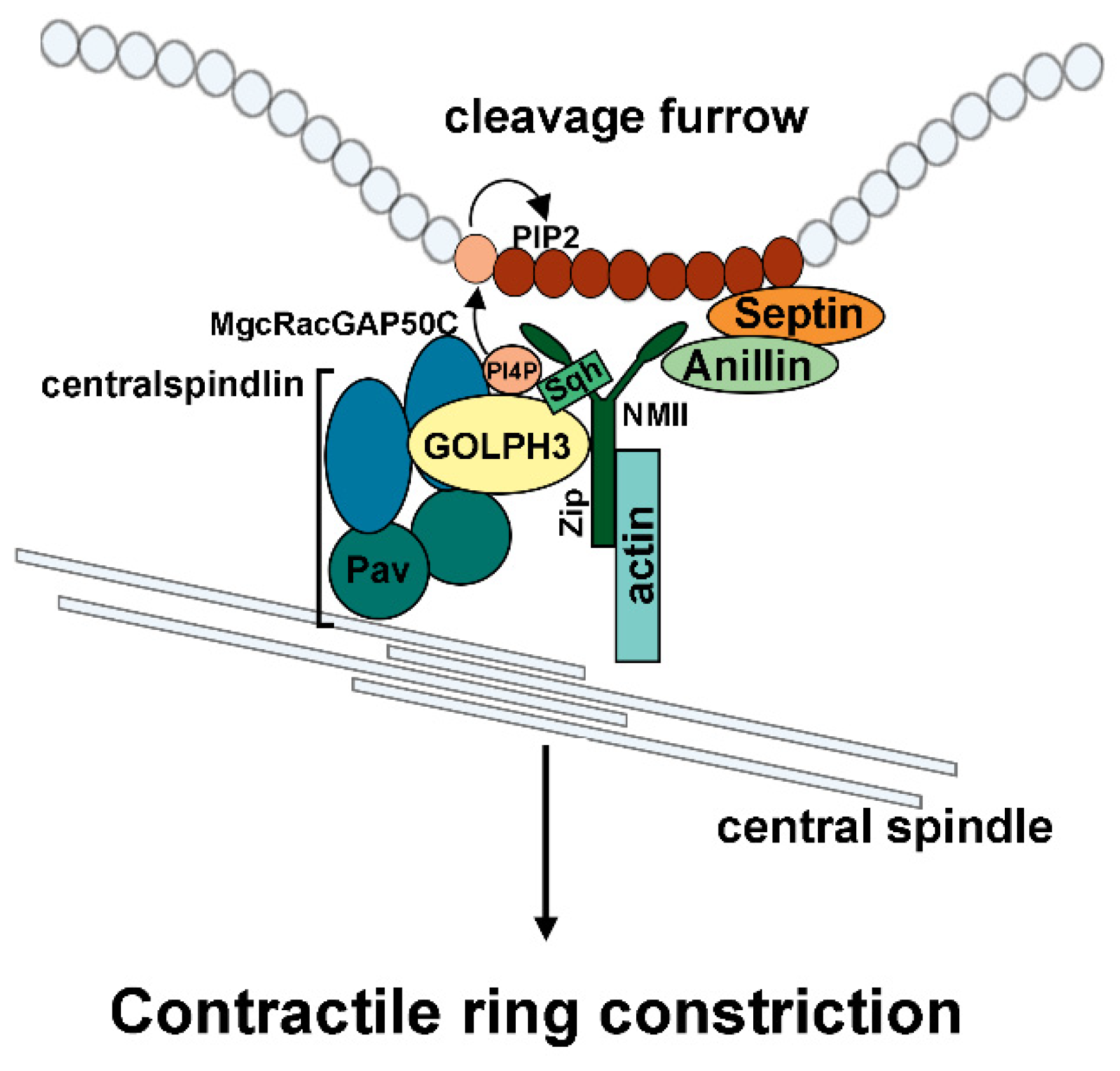
4. GOLPH3 Plays an Essential Function During Cytokinesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, C.C.; Taylor, R.S.; Lane, D.R.; Ladinsky, M.S.; Weisz, J.A.; Howell, K.E. GMx33: A novel family of trans-Golgi proteins identified by proteomics. Traffic 2000, 1, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.W.; Ward, M.A.; Blackstock, W.P.; Freeman, H.N.; Choudhary, J.S.; Lewis, A.P.; Chotai, D.; Fazel, A.; Gushue, J.N.; Paiement, J.; et al. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 2001, 276, 5112–5165. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Mardones, G.A.; Ladinsky, M.S.; Howell, K.E. GMx33 associates with the trans-Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol. Biol. Cell 2006, 17, 511–524. [Google Scholar] [CrossRef]
- Dippold, H.C.; Field, S.J. GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.S.; Schmitz, K.R.; Bessman, N.J.; Setty, T.G.; Ferguson, K.M.; Burd, C.G. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J. Cell Biol. 2009, 187, 967–975. [Google Scholar] [CrossRef]
- Sechi, S.; Colotti, G.; Belloni, G.; Mattei, V.; Frappaolo, A.; Raffa, G.D.; Fuller, M.T.; Giansanti, M.G. GOLPH3 is essential for contractile ring formation and Rab11 localization to the cleavage site during cytokinesis in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004305. [Google Scholar] [CrossRef]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef]
- Xie, M.W.; Jin, F.; Hwang, H.; Hwang, S.; Anand, V.; Duncan, M.C.; Huang, J. Insights into TOR function and rapamycin response: Chemical genomic profiling by using a high-density cell array method. Proc. Natl. Acad. Sci. USA 2005, 102, 7215–7220. [Google Scholar] [CrossRef]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Kuna, R.S.; Field, S.J. GOLPH3: A Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res. 2019, 60, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Pan, H.; Wei, W.; Yang, H.; Liu, J.; Yang, R. GOLPH3: A novel biomarker that correlates with poor survival and resistance to chemotherapy in breast cancer. Oncotarget 2017, 8, 105155–105169. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Lin, H.; Zhao, X.; Liu, G.; Wang, X.; Xu, R.; Chen, K.; Li, J.; Song, L. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 2012, 18, 4059–4069. [Google Scholar] [CrossRef]
- Wang, J.H.; Chen, X.T.; Wen, Z.S.; Zheng, M.; Deng, J.M.; Wang, M.Z.; Lin, H.X.; Chen, K.; Li, J.; Yun, J.P.; et al. High expression of GOLPH3 in esophageal squamous cell carcinoma correlates with poor prognosis. PLoS ONE 2012, 7, e45622. [Google Scholar] [CrossRef]
- Lu, M.; Tian, Y.; Yue, W.M.; Li, L.; Li, S.H.; Qi, L.; Hu, W.S.; Gao, C.; Si, L.B.; Tian, H. GOLPH3, a good prognostic indicator in early-stage NSCLC related to tumor angiogenesis. Asian Pac. J. Cancer Prev. 2014, 15, 5793–5798. [Google Scholar] [CrossRef]
- Tang, S.; Yang, R.; Zhou, X.; Pan, H.; Liu, J. Expression of GOLPH3 in patients with non-small cell lung cancer and xenografts models. Oncol. Lett. 2018, 15, 7555–7562. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, M.; Han, B. GOLPH3 high expression predicts poor prognosis in patients with resected non-small cell lung cancer: An immunohistochemical analysis. Tumour Biol. 2014, 35, 10833–10839. [Google Scholar] [CrossRef]
- Li, H.; Guo, L.; Chen, S.W.; Zhao, X.H.; Zhuang, S.M.; Wang, L.P.; Song, L.B.; Song, M. GOLPH3 overexpression correlates with tumor progression and poor prognosis in patients with clinically N0 oral tongue cancer. J. Transl. Med. 2012, 10, 168. [Google Scholar] [CrossRef]
- Hua, X.; Yu, L.; Pan, W.; Huang, X.; Liao, Z.; Xian, Q.; Fang, L.; Shen, H. Increased expression of Golgi phosphoprotein-3 is associated with tumor aggressiveness and poor prognosis of prostate cancer. Diagn. Pathol. 2012, 7, 127. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, T.; Qin, R.; Yan, Y.; Chen, C.; Chen, Y.; Yu, H.; Xia, C.; Lu, Y.; Ding, X.; et al. Overexpression of Golgi phosphoprotein-3 (GOLPH3) in glioblastoma multiforme is associated with worse prognosis. J. Neurooncol. 2012, 110, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Liu, W.; Chen, S.F.; Zhang, L.Q.; Li, X.G.; Wang, L.X. Expression of the Golgi phosphoprotein-3 gene in human gliomas: A pilot study. J. Neurooncol. 2011, 105, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zappa, F.; Failli, M.; De Matteis, M.A. The Golgi complex in disease and therapy. Curr. Opin. Cell Biol. 2018, 50, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Alterations of Golgi Structural Proteins and Glycosylation Defects in Cancer. Front. Cell Dev. Biol. 2021, 9, 665289. [Google Scholar] [CrossRef]
- Lee, Z.Y.; Lee, W.H.; Lim, J.S.; Ali, A.A.A.; Loo, J.S.E.; Wibowo, A.; Mohammat, M.F.; Foo, J.B. Golgi apparatus targeted therapy in cancer: Are we there yet? Life Sci. 2024, 352, 122868. [Google Scholar] [CrossRef]
- Halberg, N.; Sengelaub, C.A.; Navrazhina, K.; Molina, H.; Uryu, K.; Tavazoie, S.F. PITPNC1 Recruits RAB1B to the Golgi Network to Drive Malignant Secretion. Cancer Cell 2016, 29, 339–353. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. [Google Scholar] [CrossRef]
- Tu, L.; Banfield, D.K. Localization of Golgi-resident glycosyltransferases. Cell Mol. Life Sci. 2010, 67, 29–41. [Google Scholar] [CrossRef]
- Banfield, D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005264. [Google Scholar] [CrossRef]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Liu, J.; Li, S.; Setty, T.G.; Wood, C.S.; Burd, C.G.; Ferguson, K.M. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 2008, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Chen, L.; Banfield, D.K. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic 2012, 13, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Chachadi, V.B.; Petrosyan, A.; Cheng, P.W. Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of core 2 N-acetylglucosaminyltransferase 1. J. Biol. Chem. 2012, 287, 39564–39577. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.A.; Pu, H.X.; Goh, H.; Song, Z. Golgi phosphoprotein 3 mediates the Golgi localization and function of protein O-linked mannose β-1,2-N-acetlyglucosaminyltransferase 1. J. Biol. Chem. 2014, 289, 14762–14770. [Google Scholar] [CrossRef]
- Eckert, E.S.; Reckmann, I.; Hellwig, A.; Röhling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–31329. [Google Scholar] [CrossRef]
- Henry, M.D.; Campbell, K.P. Dystroglycan inside and out. Curr. Opin. Cell Biol. 1999, 11, 602–607. [Google Scholar] [CrossRef]
- Chang, W.L.; Chang, C.W.; Chang, Y.Y.; Sung, H.H.; Lin, M.D.; Chang, S.C.; Chen, C.H.; Huang, C.W.; Tung, K.S.; Chou, T.B. The Drosophila GOLPH3 homolog regulates the biosynthesis of heparan sulfate proteoglycans by modulating the retrograde trafficking of exostosins. Development 2013, 140, 2798–2807. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, R.; Lin, H.; Wang, H. Insights into the molecular regulatory network of pathomechanisms in osteochondroma. J. Cell Biochem. 2019, 120, 16362–16369. [Google Scholar] [CrossRef]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef]
- Sechi, S.; Karimpour-Ghahnavieh, A.; Frappaolo, A.; Di Francesco, L.; Piergentili, R.; Schininà, E.; D’Avino, P.P.; Giansanti, M.G. Identification of GOLPH3 Partners in Drosophila Unveils Potential Novel Roles in Tumorigenesis and Neural Disorders. Cells 2021, 10, 2336. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Russo, D.; Kurokawa, K.; Sahu, P.; Lombardi, B.; Supino, D.; Zhukovsky, M.A.; Vocat, A.; Pothukuchi, P.; Kunnathully, V.; et al. Golgi maturation-dependent glycoenzyme recycling controls glycosphingolipid biosynthesis and cell growth via GOLPH3. EMBO J. 2021, 40, e107238. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, F.M.; Martínez-Koteski, N.; Cavieres, V.A.; Mardones, G.A.; Fidelio, G.D.; Vilcaes, A.A.; Daniotti, J.L. Golgi Phosphoprotein 3 Regulates the Physical Association of Glycolipid Glycosyltransferases. Int. J. Mol. Sci. 2022, 23, 10354. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef]
- Welch, L.G.; Peak-Chew, S.Y.; Begum, F.; Stevens, T.J.; Munro, S. GOLPH3 and GOLPH3L are broad-spectrum COPI adaptors for sorting into intra-Golgi transport vesicles. J. Cell Biol. 2021, 220, e202106115. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Khorami-Sarvestani, S.; Hanash, S.M.; Fahrmann, J.F.; León-Letelier, R.A.; Katayama, H. Glycosylation in cancer as a source of biomarkers. Expert Rev. Proteom. 2024, 21, 345–365. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein Glycosylation in Cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef]
- Dobie, C.; Skropeta, D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2021, 124, 76–90. [Google Scholar] [CrossRef]
- Sethi, M.K.; Thaysen-Andersen, M.; Smith, J.T.; Baker, M.S.; Packer, N.H.; Hancock, W.S.; Fanayan, S. Comparative N-glycan profiling of colorectal cancer cell lines reveals unique bisecting GlcNAc and α-2,3-linked sialic acid determinants are associated with membrane proteins of the more metastatic/aggressive cell lines. J. Proteome Res. 2014, 13, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.R.; Shaikh, F.M.; Lucas, J.A., 4th; Lucas, J.A., 3rd; Bellis, S.L. ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J. Ovarian Res. 2008, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Kemmner, W.; Grigull, S.; Schlag, P.M. Cell surface alpha 2,6 sialylation affects adhesion of breast carcinoma cells. Exp. Cell Res. 2002, 276, 101–110. [Google Scholar] [CrossRef]
- Peng, W.; Goli, M.; Mirzaei, P.; Mechref, Y. Revealing the Biological Attributes of N-Glycan Isomers in Breast Cancer Brain Metastasis Using Porous Graphitic Carbon (PGC) Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). J. Proteome Res. 2019, 18, 3731–3740. [Google Scholar] [CrossRef]
- Li, Q.; Li, G.; Zhou, Y.; Zhang, X.; Sun, M.; Jiang, H.; Yu, G. Comprehensive N-Glycome Profiling of Cells and Tissues for Breast Cancer Diagnosis. J. Proteome Res. 2019, 18, 2559–2570. [Google Scholar] [CrossRef]
- Isaji, T.; Im, S.; Gu, W.; Wang, Y.; Hang, Q.; Lu, J.; Fukuda, T.; Hashii, N.; Takakura, D.; Kawasaki, N.; et al. An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J. Biol. Chem. 2014, 289, 20694–20705. [Google Scholar] [CrossRef]
- Gu, J.; Isaji, T. Specific sialylation of N-glycans and its novel regulatory mechanism. Glycoconj. J. 2024, 41, 175–183. [Google Scholar] [CrossRef]
- Clayton, E.L.; Minogue, S.; Waugh, M.G. Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Prog. Lipid Res. 2013, 52, 294–304. [Google Scholar] [CrossRef]
- Morrow, A.A.; Alipour, M.A.; Bridges, D.; Yao, Z.; Saltiel, A.R.; Lee, J.M. The lipid kinase PI4KIIIβ is highly expressed in breast tumors and activates Akt in cooperation with Rab11a. Mol. Cancer Res. 2014, 12, 1492–1508. [Google Scholar] [CrossRef]
- Berditchevski, F.; Tolias, K.F.; Wong, K.; Carpenter, C.L.; Hemler, M.E. A novel link between integrins, transmembrane-4 superfamily proteins (CD63 and CD81), and phosphatidylinositol 4-kinase. J. Biol. Chem. 1997, 272, 2595–2598. [Google Scholar] [CrossRef]
- Yauch, R.L.; Berditchevski, F.; Harler, M.B.; Reichner, J.; Hemler, M.E. Highly stoichiometric, stable, and specific association of integrin alpha3beta1 with CD151 provides a major link to phosphatidylinositol 4-kinase, and may regulate cell migration. Mol. Biol. Cell 1998, 9, 2751–2765. [Google Scholar] [CrossRef]
- Yauch, R.L.; Kazarov, A.R.; Desai, B.; Lee, R.T.; Hemler, M.E. Direct extracellular contact between integrin alpha(3)beta(1) and TM4SF protein CD151. J. Biol. Chem. 2000, 275, 9230–9238. [Google Scholar] [CrossRef] [PubMed]
- Isaji, T.; Im, S.; Kameyama, A.; Wang, Y.; Fukuda, T.; Gu, J. A complex between phosphatidylinositol 4-kinase IIα and integrin α3β1 is required for N-glycan sialylation in cancer cells. J. Biol. Chem. 2019, 294, 4425–4436. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Isaji, T.; Oyama, Y.; Xu, X.; Liu, J.; Hanamatsu, H.; Yokota, I.; Miura, N.; Furukawa, J.I.; Fukuda, T.; et al. Focal-adhesion kinase regulates the sialylation of N-glycans via the PI4KIIα-PI4P pathway. J. Biol. Chem. 2023, 299, 105051. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef]
- Stocker, H.; Radimerski, T.; Schindelholz, B.; Wittwer, F.; Belawat, P.; Daram, P.; Breuer, S.; Thomas, G.; Hafen, E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat. Cell Biol. 2003, 5, 559–565. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Scott, K.L.; Chin, L. Signaling from the Golgi: Mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis. Clin. Cancer Res. 2010, 16, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, P.; Houghton, F.J.; McMillan, P.J.; Hanssen, E.; Gleeson, P.A. The Golgi ribbon in mammalian cells negatively regulates autophagy by modulating mTOR activity. J. Cell Sci. 2018, 131, jcs211987. [Google Scholar] [CrossRef]
- Hao, F.; Kondo, K.; Itoh, T.; Ikari, S.; Nada, S.; Okada, M.; Noda, T. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 2018, 131, jcs208017. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, X.F. Endoplasmic reticulum and Golgi localization sequences for mammalian target of rapamycin. Mol. Biol. Cell 2007, 18, 1073–1082. [Google Scholar] [CrossRef]
- Manifava, M.; Smith, M.; Rotondo, S.; Walker, S.; Niewczas, I.; Zoncu, R.; Clark, J.; Ktistakis, N.T. Dynamics of mTORC1 activation in response to amino acids. eLife 2016, 5, e19960. [Google Scholar] [CrossRef]
- Thomas, J.D.; Zhang, Y.J.; Wie, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Cesare, G.; Sechi, S.; Fraschini, R.; Vaccari, T.; Giansanti, M.G. GOLPH3 protein controls organ growth by interacting with TOR signaling proteins in Drosophila. Cell Death Dis. 2022, 13, 1003. [Google Scholar] [CrossRef]
- Frappaolo, A.; Giansanti, M.G. Using Drosophila melanogaster to Dissect the Roles of the mTOR Signaling Pathway in Cell Growth. Cells 2023, 12, 2622. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Chern, J.J.; Cai, Y.; Liu, M.; Choi, K.W. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature 2007, 445, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Nam, B.Y.; Li, J.J.; Park, J.T.; Lee, S.H.; Kim, D.H.; Kim, J.Y.; Kang, H.Y.; Han, S.H.; Yoo, T.H.; et al. Translationally controlled tumour protein is associated with podocyte hypertrophy in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 1205–1217. [Google Scholar] [CrossRef]
- Le, T.P.; Vuong, L.T.; Kim, A.R.; Hsu, Y.C.; Choi, K.W. 14-3-3 proteins regulate Tctp-Rheb interaction for organ growth in Drosophila. Nat. Commun. 2016, 7, 11501. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Wang, T.; Blumhagen, R.; Lao, U.; Kuo, Y.; Edgar, B.A. LST8 regulates cell growth via target-of-rapamycin complex 2 (TORC2). Mol. Cell Biol. 2012, 32, 2203–2213. [Google Scholar] [CrossRef]
- Hwang, Y.; Kim, L.C.; Song, W.; Edwards, D.N.; Cook, R.S.; Chen, J. Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth. In. Vivo. Cancer Res. 2019, 79, 3178–3184. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Fraschini, R.; Capalbo, L.; Gottardo, M.; Belloni, G.; Glover, D.M.; Wainman, A.; Giansanti, M.G. Rab1 interacts with GOLPH3 and controls Golgi structure and contractile ring constriction during cytokinesis in Drosophila melanogaster. Open Biol. 2017, 7, 160257. [Google Scholar] [CrossRef]
- D’Avino, P.P.; Giansanti, M.G.; Petronczki, M. Cytokinesis in animal cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a015834. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Fraschini, R.; Giansanti, M.G. A novel coordinated function of Myosin II with GOLPH3 controls centralspindlin localization during cytokinesis in Drosophila. J. Cell Sci. 2020, 133, jcs252965. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Sauri-Nadal, T.; Menendez, O.J.; Oliveras-Ferraros, C.; Cufí, S.; Corominas-Faja, B.; López-Bonet, E.; Menendez, J.A. Ser2481-autophosphorylated mTOR colocalizes with chromosomal passenger proteins during mammalian cell cytokinesis. Cell Cycle 2012, 11, 4211–4221. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Hosea, R.; Hillary, S.; Naqvi, S.; Wu, S.; Kasim, V. The two sides of chromosomal instability: Drivers and brakes in cancer. Signal Transduct. Target Ther. 2024, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.; D’Avino, P.P. Investigating cytokinesis failure as a strategy in cancer therapy. Oncotarget 2016, 7, 87323–87341. [Google Scholar] [CrossRef]
- Joshi, S.; Braithwaite, A.W.; Robinson, P.J.; Chircop, M. Dynamin inhibitors induce caspase-mediated apoptosis following cytokinesis failure in human cancer cells and this is blocked by Bcl-2 overexpression. Mol. Cancer 2011, 10, 78. [Google Scholar] [CrossRef]
- Luwor, R.; Morokoff, A.P.; Amiridis, S.; D’Abaco, G.; Paradiso, L.; Stylli, S.S.; Nguyen, H.P.T.; Tarleton, M.; Young, K.A.; O’Brien, T.J.; et al. Targeting Glioma Stem Cells by Functional Inhibition of Dynamin 2: A Novel Treatment Strategy for Glioblastoma. Cancer Invest. 2019, 37, 144–155. [Google Scholar] [CrossRef]
- Cilibrasi, C.; Guzzi, A.; Bazzoni, R.; Riva, G.; Cadamuro, M.; Hochegger, H.; Bentivegna, A. A Ploidy Increase Promotes Sensitivity of Glioma Stem Cells to Aurora Kinases Inhibition. J. Oncol. 2019, 2019, 9014045. [Google Scholar] [CrossRef]
- Chen, A.S.; Wardwell-Ozgo, J.; Shah, N.N.; Wright, D.; Appin, C.L.; Vigneswaran, K.; Brat, D.J.; Kornblum, H.I.; Read, R.D. Drak/STK17A Drives Neoplastic Glial Proliferation through Modulation of MRLC Signaling. Cancer Res. 2019, 79, 1085–1097. [Google Scholar] [CrossRef]
- Giansanti, M.G.; Piergentili, R. Linking GOLPH3 and Extracellular Vesicles Content-a Potential New Route in Cancer Physiopathology and a Promising Therapeutic Target is in Sight? Technol. Cancer Res. Treat. 2022, 21, 15330338221135724. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Chen, Y.; Jin, S.; Gao, Y.; Zhang, D.; Wu, Y. Evaluation of the Oncogene Function of GOLPH3 and Correlated Regulatory Network in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 669684. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiang, S.; Huang, A.; Gao, Y.; Peng, B.; Li, Z.; Ma, W.; Songyang, Z.; Zhang, S.; He, M.; et al. GOLPH3 Promotes Cancer Growth by Interacting with STIP1 and Regulating Telomerase Activity in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2020, 10, 575358. [Google Scholar] [CrossRef] [PubMed]
- Farber-Katz, S.E.; Dippold, H.C.; Buschman, M.D.; Peterman, M.C.; Xing, M.; Noakes, C.J.; Tat, J.; Ng, M.M.; Rahajeng, J.; Cowan, D.M.; et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 2014, 156, 413–427. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frappaolo, A.; Zaccagnini, G.; Giansanti, M.G. GOLPH3-mTOR Crosstalk and Glycosylation: A Molecular Driver of Cancer Progression. Cells 2025, 14, 439. https://doi.org/10.3390/cells14060439
Frappaolo A, Zaccagnini G, Giansanti MG. GOLPH3-mTOR Crosstalk and Glycosylation: A Molecular Driver of Cancer Progression. Cells. 2025; 14(6):439. https://doi.org/10.3390/cells14060439
Chicago/Turabian StyleFrappaolo, Anna, Gianluca Zaccagnini, and Maria Grazia Giansanti. 2025. "GOLPH3-mTOR Crosstalk and Glycosylation: A Molecular Driver of Cancer Progression" Cells 14, no. 6: 439. https://doi.org/10.3390/cells14060439
APA StyleFrappaolo, A., Zaccagnini, G., & Giansanti, M. G. (2025). GOLPH3-mTOR Crosstalk and Glycosylation: A Molecular Driver of Cancer Progression. Cells, 14(6), 439. https://doi.org/10.3390/cells14060439