1. Introduction
Stroke, the second leading cause of mortality worldwide, is responsible for approximately 5.5 million deaths annually and continues to be a significant contributor to global disability [
1]. Stroke is primarily classified into hemorrhagic and ischemic subtypes, with ischemic strokes accounting for up to 71% [
2]. Following the impact of ischemia, a cascade of interconnected cytoplasmic and nuclear events occurs at the damaged site, leading to bioenergetic depletion, excitotoxicity, excessive calcium influx, oxidative stress, and inflammatory responses that ultimately result in neuronal cell death [
3]. The inflammation caused by cell death can further aggravate the injury by impairing the ischemic penumbra through bystander effects. The mounting evidence suggests that post-ischemic inflammation plays a pivotal role in the progression of brain injury. The severity of outcomes in acute ischemic stroke is significantly influenced by the magnitude of this inflammatory response [
4,
5]. The appropriate termination of neuroinflammation after a stroke has been demonstrated to effectively promote tissue repair and regeneration, thereby offering considerable therapeutic benefits [
6].
The microglia, as the resident immune cells of the central nervous system [
7], are indispensable inflammatory cells that contribute to maintaining homeostasis and play an important role in neuroinflammation and neurodegenerative diseases [
8]. Following the onset of ischemic stroke, microglia can promote tissue repair through the clearance of damaged cells or debris and the production of anti-inflammatory cytokines and growth factors [
9,
10]. The cells can also release proinflammatory cytokines, such as IL-6, IL-1β, and iNOS, concurrently contributing to the inflammatory response [
11,
12,
13]. The central role of microglia in neuroinflammation renders them a viable therapeutic target for modulating post-ischemic stroke inflammation [
14]. The NF-κB signaling pathway is a canonical cytokine-mediated signaling cascade that plays a crucial role in inflammatory processes [
15]. Consequently, the inhibition of NF-κB activation may represent a potential strategy for mitigating inflammation and safeguarding against brain damage.
The process of autophagy plays a pivotal role in the innate immune response and functions as an adaptive mechanism for cells to adapt to metabolic stress and environmental fluctuations [
16,
17]. It facilitates the degradation or elimination of damaged proteins and organelles within the cytoplasm through the lysosomal degradation pathway, thereby ensuring cellular homeostasis. When triggered by diverse stress conditions, autophagy safeguards cell survival and homeostasis by clearing protein aggregates and dysfunctional mitochondria, recovering amino acids, fatty acids, and glucose to sustain energy balance while mitigating inflammation [
18]. The mild-to-moderate activation of autophagy can, thus, serve as a cellular pro-survival mechanism. However, excessive or prolonged autophagic activation may ultimately result in cell death [
19]. The excessive accumulation of autophagosomes may necessitate the exacerbation of cellular component degradation, leading to cellular injury and the increased secretion of inflammatory factors [
20,
21,
22]. Therefore, exploring autophagy in the context of ischemic stroke challenges may provide innovative strategies for stroke prevention and treatment.
Autophagy and inflammation are two critical, interconnected processes by which microglia execute their functions during ischemic stroke. Under normal circumstances, autophagy is considered to exert a regulatory effect on microglial inflammation; however, under activated conditions, it may serve a dual role as both an inhibitory and stimulatory factor in ischemic inflammation [
23]. The application of autophagy inhibitors, for instance, enhances the expression of inflammatory genes in adipocytes; conversely, the activation of autophagy reduces the expression of inflammatory genes, thereby indicating that autophagy has the capacity to modulate inflammatory responses [
24]. The use of autophagy inhibitors, such as 3-methyladenine, conversely impedes autophagic activity and alleviates LPS-induced acute lung injury by suppressing inflammation and reversing autophagy. This suggests that the inhibition of autophagy may also mitigate inflammatory responses [
25]. The effective regulation of the interplay between microglial autophagy and inflammation may offer novel avenues for research aimed at enhancing protection against ischemic stroke.
The novel compound SHPL-49, developed by our team as a structurally modified derivative of salidroside, has exhibited neuroprotective effects against ischemic stroke [
26,
27,
28]. The pharmacological effects of SHPL-49 were initially discovered to exert neuroprotective properties through the inhibition of calcium influx, oxidative stress injury, apoptosis, and other mechanisms. This subsequently leads to a reduction in cerebral infarction volume and effectively alleviates ischemic brain injury [
27]. After further investigation into the mechanism of action and potential targets of SHPL-49, we have discovered that SHPL-49 exhibits the ability to enhance neuronal survival and alleviate acute ischemic stroke by facilitating the NR2A-CAMKIIα-Akt/CREB pathway [
28]. Moreover, our findings indicate that SHPL-49 can activate angiogenesis via the microglia-mediated VEGFR2/Akt/eNOS pathway and suppress the p38 MAPK/MMP-9 signaling cascade to mitigate blood–brain barrier (BBB) disruption in microglia, thereby exerting neuroprotective effects [
29]. These results indicate that microglia potentially exert a crucial influence on regulating the neuroprotective effects of SHPL-49 in cases of ischemic stroke. Additionally, our study demonstrated that the regulation of microglia by SHPL-49 induces the secretion of neurotrophic factors while reducing the release of inflammatory mediators. Ultimately, SHPL-49 attenuates neuronal damage caused by ischemia and promotes neurogenesis through the activation of the BDNF/TrkB/Gap43 pathway, thereby enhancing synaptic plasticity [
26]. Although our study demonstrated the potential of SHPL-49 in mitigating microglia-induced neuroinflammation, the precise underlying mechanisms remain elusive and warrant further investigation. In light of the pivotal regulatory roles played by microglia and autophagy in the neuroinflammatory response triggered by ischemic stroke, this study aims to investigate whether SHPL-49 can ameliorate the occurrence of neuroinflammation through the modulation of microglial autophagy, thereby conferring neuronal protection. This may offer a promising avenue for the development and clinical application of drugs targeting ischemic stroke.
2. Materials and Methods
2.1. Reagents
SHPL-49 was produced by Shanghai Hutchison Pharmaceuticals, lot number 3001430101. Edaravone was purchased from J&K Scientific Co., Ltd. (Beijing, China).
2.2. Animals
Male SD rats weighing 260–280 g were procured from Beijing Weitong Lihua Laboratory Animal Technology Co., Ltd. (Beijing, China). Male rats were chosen for the experiment because the androgen environment in males is relatively stable and does not fluctuate with the physiological cycle, which makes the experimental conditions easy to control. All the experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine with the ethics number PZSHUTCM2303220004. All the experiments were conducted in accordance with The Guide for The Care and Use of Laboratory Animals published by the National Institutes of Health Animals, and ARRIVE guidelines were followed. The rats were housed in a temperature-controlled chamber maintained at 22–26 °C with relative humidity between 40% and 70% under a 12 h light/dark cycle. The rats were provided with unrestricted access to water and food.
2.3. Establishment of Rat Model of Permanent Middle Cerebral Artery Occlusion (pMCAO)
The rats were positioned on a heating pad and subjected to anesthesia using an induction dose of 3% and a maintenance dose of 2.5% isoflurane, ensuring body temperature was maintained at 37 °C ± 0.5 °C [
30]. The left common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were surgically exposed through a cervical incision. The proximal and distal CCA were ligated, while the distal ECA was also ligated. A silica-coated wire (Guangzhou Jialing Biotechnology Co., Ltd., Guangzhou, China) was carefully inserted through a small incision in the CCA and gradually advanced into the anterior end of the middle cerebral artery. The wound was sutured and disinfected after occlusion. During ligation, alterations in cerebral blood flow were monitored using a laser Doppler flow meter (Moor VSM, Axminster, UK). Success was defined as a reduction of 70% in ipsilateral cerebral blood flow compared to the contralateral hemisphere [
27]. The survival rate of pMCAO rats was 80%. The physical and mental states of rats were closely monitored on a daily basis throughout the experiment. SHPL-49 and ED were administered via intravenous injection through the tail vein. After the completion of the drug intervention, the experimental rats were euthanized by bleeding from the abdominal aorta under deep anesthesia.
2.4. Cell Culture
The BV2 cells of microglia from mice were cultured in complete DMEM (HAKATA, China, A19008) supplemented with 10% fetal bovine serum (FBS) obtained from Hyclone, UT, USA, SH30071, and 1% penicillin–streptomycin. The highly differentiated PC-12 cells were acquired from Shanghai Zhong Qiao Xin Zhou Biotech Co., Ltd., Beijing, China. and cultured in complete DMEM. Each type of cell was cultured under a humidified atmosphere containing 5% CO2 at a temperature of 37 °C.
2.5. Establishment of Cell Model for Simulating Oxygen–Glucose Deprivation
The oxygen–glucose deprivation (OGD) technique was employed to simulate cerebral ischemia in cellular models. After washing the cells with phosphate-buffered saline (PBS) (HAKATA, Shanghai, China, A19712), the culture medium was substituted with sugar-free and serum-free 1640 medium (HAKATA, Shanghai, China, A19011) in the Model group. The drug administration groups were provided with a sugar-free, serum-free medium containing the drug instead of the media. The hypoxic tank was connected to a hypoxic apparatus to achieve hypoxia, and the gas composition was altered to 95% N2, 4% CO2, and 1% O2. Subsequently, the hypoxic tank was placed inside the cell culture chamber (Thermo, 4111FO, Waltham, MA, USA).
2.6. Establishment of Co-Culture Model Integrating BV2 Cells and PC-12 Cells
A co-culture cell model of BV2 and PC12 cells was established, with BV2 cells placed on the top layer of Transwell chambers (LABSELCT, Hefei, Anhui, China, 32023185X). Simultaneously, PC-12 cells within the chambers were maintained in DMEM and co-cultured for 2 days to ensure stability. Subsequently, the co-culture system was subjected to an OGD environment for further experiments.
2.7. Immunofluorescent Staining Assay
Immunofluorescence staining of brain sections: The brain paraffin sections were dewaxed and subjected to a gradient rehydration process using xylene and ethanol. Subsequently, they were blocked with 10% goat serum for 1 h at room temperature. The sections were subsequently incubated overnight at 4 °C with the designated primary antibodies as follows: P62/SQSTM1 (1:100, Boster Biological Technology, Wuhan, China, BM4385), LC3B (1:200, AiFang biologic, Changsha, Hunan, China, AF11004), and Iba1 (1:500, Servicebio, Wuhan, China, GB12105-100). After being washed with PBS, the sections were incubated for 2 h at room temperature with secondary antibodies against goat anti-mouse IgG H&L (Alexa Fluor
® 488) (1:500, Abcam, Cambridge, UK, ab150113) or donkey anti-rabbit IgG H&L (Alexa Fluor
® 555) (1:500, Abcam, Cambridge, UK, ab150074). After washing three times with PBS (Servicebio, Wuhan, China, G0002), the sections were treated with an anti-fluorescence quenching sealer (Beyotime, Shanghai, China, P0131) containing 4, 6-diamidino-2-phenylindole (DAPI) and covered with coverslips. Tissue sections were viewed using a laser confocal microscope (Nikon, Ti2, Otawara, Japan) [
31]. Immunofluorescence staining of BV2 cells: The medium was discarded, and the cells were fixed with a 4% paraformaldehyde solution. Subsequently, the cells were permeated with 0.5% Triton X-100 (Adamas-beta
®, Shanghai, China) and subsequently incubated with 5% bovine serum albumin (BSA, Beyotime, Shanghai, China) at room temperature for 30 min. After washing, the cells were incubated overnight at 4 °C with the subsequent primary antibodies, each of which possesses specific characteristics as described below: LC3 (1:1000, AiFang biologic, Hunan, China, AF11004) and NF-κB p65 (1:400, Cell Signaling Technology, Danvers, MA, USA, D14E12). After washing, the cells were incubated for 1 h at room temperature with goat anti-mouse IgG H&L (Alexa Fluor
® 488) (1:500, Abcam, Cambridge, UK, ab150113) or donkey anti-rabbit IgG H&L (Alexa Fluor
® 555) (1:500, Abcam, Cambridge, UK, ab150074) secondary antibodies. The nuclei were stained with DAPI and subsequently observed under a laser confocal microscope (Nikon, Otawara, Japan, Ti2).
2.8. Immunohistochemistry
Rat brain tissue sections were subjected to dewaxing and dehydration, followed by antigen retrieval using an antigen retrieval solution. After rinsing three times with PBS (Servicebio, Wuhan, China, G0002), the sections were incubated with 5% BSA (Beyotime, Shanghai, China, ST023) for one hour at room temperature. Subsequently, the sections were incubated overnight at 4 °C with primary antibodies against NF-κB (1:400, Cell Signaling Technology, MA, USA, D14E12) and IL-6 (Servicebio, Wuhan, China, GB11117-100). The following day, the sections were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H + L) secondary antibody at room temperature for one hour. Diaminobenzidine was used for color development, and cell nuclei were counterstained with Harris hematoxylin. The sections were examined under an inverted fluorescence microscope (Nikon) using white light illumination. A quantitative analysis of the images was performed using ImageJ 1.8.0 software. For statistical analysis, at least three sections were randomly selected from each group, and 3–5 different regions were randomly chosen from each section.
2.9. Real-Time Quantitative PCR (RT-qPCR)
The total mRNA from BV2 cells was extracted using Trizol (Vazyme, Nanjing, Jiangsu, China, R401-01), and the concentrations of mRNA were determined using an ultramicro spectrophotometer (DeNovix, Wilmington, DE, USA, TFB-TIWI-01). After the removal of DNA and reverse transcription to generate the cDNA (Vazyme, Jiangsu, China, R223-01), real-time quantitative PCR (RT-qPCR) was performed using Vazyme’s kit (Vazyme, Jiangsu, China, R711-03). The data were analyzed by employing the 2
−ΔΔct method [
32]. The PCR primers utilized in the experiment were as follows. β-actin: F: GGC-TGTATTCCCCTCCATCG, R: CCAGTTGGT-AACAATGCCATGT; IL-6: F: CTCTGCAAGAGACTTCCATCCAGT, R: CATTTCCACGATTTCCCAGAGA; IL-1β: F: CCTGTCCTGCGTGTTGAAAGA, R: GGGAACTGGGCAGACTCAAA;
iNOS: F: GGTATGCTGTGTT-TGGCCTT, R: GCAGCCTCTTGTCTTTGACC.
2.10. ELISA: Enzyme-Linked Immunosorbent Assay
The protein levels of the inflammatory factors IL-6 (Boster Biological Technology, Wuhan, China, EK0411), IL-1β (Boster Biological Technology, Wuhan, China, EK0394), and iNOS (JONLNBIO, Shanghai, China, JL20675) in the supernatants of treated BV2 cells were quantified using ELISA kits as per the manufacturer’s instructions.
2.11. Hoechst 33342 Staining Assay
The working solution of Hoechst 33342 (Servicebio, Wuhan, China, G1127) staining was added to the cell cultures at a volume of approximately one-tenth of the medium, ensuring the full coverage of the sample to be stained. The cultures were then incubated for 15 min in a 37 °C incubator. After the removal of the Hoechst 33342 staining working solution, the cell plates were washed 3 times with PBS and observed under a laser confocal microscope (Nikon, Otawara, Japan, Ti2).
2.12. Western Blot Assay
To extract the cytoplasmic and nuclear proteins, cells were lysed using a nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China, P0027) following the manufacturer’s instructions. Subsequently, the lysates were subjected to ultracentrifugation at 12,000× g for 10 min at 4 °C, and the resulting supernatants were collected as the cytoplasmic fraction. The pelleted nuclei were resuspended in a buffer containing 1 mM phenyl methane sulfonyl fluoride (PMSF, Beyotime, Shanghai, China, ST505). After incubation at 4 °C for 30 min, the lysates were centrifuged, and the supernatants, which contained the nuclear proteins, were stored at −80 °C. Whole-cell extracts were obtained as previously described. Total protein samples from BV2 and PC12 cells, as well as rat brain tissues, were extracted using RIPA lysis buffer (Beyotime, Shanghai, China, P0013B), and protein concentrations were measured using a BCA kit (Beyotime, Shanghai, China, P0010).
The protein samples were separated using SDS-PAGE and transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA, 1620177). Membranes were incubated with 5% nonfat milk (Beyotime, Shanghai, China, P0216) or BSA (Beyotime, Shanghai, China, ST023) followed by overnight incubation at 4 °C with the following primary antibodies: NF-κB (1:1000, Cell Signaling Technology, MA, USA, D14E12), p-IKKβ (1:1000, UpingBio, Hangzhou, Zhejiang, China, YP-Ab-14443), IKKβ (1:1000, Boster, Wuhan, China, BM4875), p-IκBα (1:1000, UpingBio, Zhejiang, China, YP-Ab-01253), IKBα (1:1000, UpingBio, Zhejiang, China, YP-Ab-01830), P62/SQSTM1 (1:1000, Boster, Wuhan, China, BM4385), LC3B (1:200, PTMab, Zhejiang, China, PTM-6384), ATG5 (1:1000, Cell Signaling Technology, MA, USA, D5F5U), Bclin1 (1:1000, Cell Signaling Technology, MA, USA, D40C5), LAMP2 (1:200, UpingBio, Zhejiang, China, YP-Ab-14094), Bax (1:1000, Cell Signaling Technology, MA, USA, 14796), Bcl-2 (1:1000, AiFang biologic, Hunan, China, AF0060), Cleaved caspase-3 (1:1000, Cell Signaling Technology, MA, USA, 9661S), and Cleaved caspase-9 (1:1000, Cell Signaling Technology, MA, USA, 9507S). The membranes were incubated with HRP-conjugated goat anti-rabbit IgG (H + L) (1:5000, Beyotime, Shanghai, China, A0208) or goat anti-mouse IgG (H + L) (1:5000, Beyotime, Shanghai, China, A0216) secondary antibodies for 2 h at room temperature. Immunolabeling was detected using ECL chemiluminescence (Meilunbio, Dalian, China, MA0186). Immunoblots were semi-quantitatively analyzed using ImageJ software (1.52a) [
33].
2.13. Cell Viability Assay
The MTT reduction assay assessed the viability of PC12 cells. Briefly, the cells were rinsed with phosphate-buffered saline (PBS, Servicebio, Wuhan, China, G0002) and then incubated with a 5 mg/mL MTT reagent for 3 h at 37 °C. After removing the medium, cell lysis was performed using 1 mL of dimethyl sulfoxide, and the absorbance was read at 540 nm using an enzyme-labeled instrument (Thermo Fisher Scientific, Waltham, MA, USA, Multiskan GO).
2.14. Analysis by Transmission Electron Microscope
The brain tissues were sectioned into 1 mm3 pieces and fixed in a solution of 2.5% glutaraldehyde for 24 h, followed by post-fixation with 1% osmium tetroxide for 3 h. Subsequently, the samples were dehydrated using an ethanol gradient (50%, 70%, 90%, and finally 100%) for a duration of 10 min each. After embedding the samples in resin, they were sliced to a thickness of approximately 50–60 nm. The resulting sections were stained with uranyl acetate and lead citrate before being examined under a transmission electron microscope (JEOL, Tokyo, Japan, JEM-F200).
2.15. Adenovirus Infection and the Quantification of mRFP-GFP-LC3 Dots
Initially, BV2 cells were cultured in 12-well plates and subsequently infected with mRFP-GFP-LC3 adenovirus (HANBIO, Shanghai, China). After infection, the cells were treated with various drugs. Nuclear staining was performed using DAPI, and visualization was achieved through confocal microscopy (Nikon, Otawara, Japan, Ti2). The localization of LC3 was monitored using the mRFP tag, and a decrease in GFP expression indicated the fusion of autophagosomes with lysosomes and the eventual formation of autolysosomes. A change in pH results in the quenching of GFP fluorescence, leaving only detectable red fluorescence. During microscopic imaging, the red and green fluorescence images are merged, with yellow dots indicating autophagosomes and red dots indicating autophagolysosomal. To quantify the intensity of autophagic flux, the number of yellow and red dots was counted.
2.16. Lyso-Tracker Green Staining Assay
The presence of lysosomes was assessed by performing Lyso-Tracker Green staining (Servicebio, Wuhan, China, G1722) following the supplier’s instructions. The cells were incubated with Lyso-Tracker Green (50 nM) at 37 °C for 30 min. Subsequently, the images were captured using confocal microscopy (Nikon, Otawara, Japan, Ti2) [
34].
2.17. Statistical Analysis
A statistical analysis of the data was performed using GraphPad Prism 9.0 software, with the results presented as the mean ± standard deviation. A one-way or two-way analysis of variance (ANOVA) was utilized for statistical testing, and a significance level of p < 0.05 was applied.
2.18. Statement of Ethics
The protocols and procedures involving animals in the experiment were all approved by the Ethics Committee of Shanghai University of Traditional Chinese Medicine after ethical review.
4. Discussion
The incidence and mortality rates of ischemic stroke are considerably high, rendering it a prevalent disease that exerts a significant impact on human health [
35]. Inflammation subsequent to an ischemic stroke extends beyond the surrounding ischemic lesions, disseminating throughout the entire brain and persisting for an extended duration. This sustained inflammatory response profoundly influences the pathological and physiological alterations in cerebral tissue following a stroke [
36].
Microglial cells, serving as the primary mediators of neuroimmune inflammation in the brain following a stroke, play pivotal roles in immune recruitment, regulation, inflammation modulation, phagocytosis facilitation, and vascular repair promotion. Therefore, they are indispensable for the development of stroke therapeutics [
37]. In this study, we demonstrated that SHPL-49 effectively inhibits neuroinflammation by modulating microglial cell autophagy and exerts significant neuroprotective effects. Our findings indicate that SHPL-49 suppresses the autophagic process in microglial cells within the brain tissue of pMCAO rats, thereby attenuating the NF-κB signaling pathway and ameliorating neuroinflammatory responses. Furthermore, cell experiments further validated the inhibitory effects of SHPL-49 on lysosomal activity in OGD-BV2 cells, as well as its ability to impede the fusion between autophagosomes and lysosomes. Ultimately, this leads to the suppression of excessive autophagy in OGD-BV2 cells, resulting in the reduced activation of the NF-κB signaling pathway and decreased protein expression levels of inflammatory factors. Additionally, by inhibiting autophagy, SHPL-49 enhances the viability of co-cultured PC12 cells, thereby contributing to its neuroprotective effects against ischemic stroke. Nevertheless, our study possesses certain limitations. Given that BV2 cells are immortalized cell lines, they cannot fully replicate the physiological and functional intricacies of primary microglial cells. Previous studies utilizing RNA-seq have compared transcriptomic alterations in BV2 microglial cell lines and primary microglia (PM) following lipopolysaccharide (LPS) stimulation [
38]. These studies revealed that PM exhibited a more robust response to LPS, with a significantly greater number of altered transcripts compared to the BV2 cell line. Consequently, future research endeavors will aim to further validate the efficacy of SHPL-49 on primary glial cells to comprehensively and accurately assess its potential as a therapeutic agent for ischemic stroke.
The NF-κB signaling pathway serves as the central mediator of inflammatory reactions, playing important roles in cellular responses to various stimuli [
39]. Upon stimulation by hypoxia, inflammatory cytokines, microbial products, and other factors, signaling molecules, including the IKK complex, become activated. This activation leads to the phosphorylation and degradation of IκB proteins, resulting in the release and activation of NF-κB. The liberated NF-κB translocates from the cytoplasm to the nucleus, where it activates the transcription of target genes such as IL-1β and IL-6, thereby augmenting the levels of inflammatory factors [
40]. In this study, we observed a significant increase in the protein levels of NF-κB in the rat pMCAO model. Additionally, the NF-κB signaling pathway was found to be activated in OGD-BV2 cells. Moreover, there was an elevation in the phosphorylation ratios of IKK and IκB-α proteins, along with a substantial rise in the protein levels of inflammatory factors IL-6, IL-1β, and iNOS. The administration of SHPL-49 significantly suppressed the activation of the NF-κB signaling pathway in rat pMCAO and OGD-BV2 cells, leading to a reduction in the expression levels of inflammatory factors. However, the precise mechanism underlying SHPL-49’s inhibition of neuroinflammation remains elusive and warrants further investigation.
Autophagy is a vital cellular process that facilitates the degradation or elimination of damaged proteins and organelles within the cytoplasm through the lysosomal degradation pathway [
41]. Autophagy represents an intricate mechanism initiated by genetic regulation involving the gradual formation of double-membrane structures. The Beclin-1/VPS34 complex subsequently facilitates the continuous elongation of autophagic double-membrane structures, while the ATG12-ATG5 complex associates with ATG16 to accomplish aggregation [
42]. The aggregated complexes fuse with autophagic vesicles to form autophagosomes. During the formation of autophagosomes, the Beclin-1 protein promotes the conversion of LC3-I, a key autophagy protein, into LC3-II [
43]. Consequently, there exists a significant correlation between the abundance of LC3-II and the quantity of autophagic bodies. However, the quantification of LC3-II at a specific point is insufficient to determine the autophagic flux, which serves as a more comprehensive indicator of overall autophagic degradation rather than solely reflecting autophagosome formation [
44]. The STX17 complex ultimately combines with SNAP29 and VAMP8 to form the SNARE complex, which subsequently translocates to the membrane of autophagic bodies. This facilitates the fusion between lysosomes and autophagic bodies, leading to the formation of autolysosomes [
45].
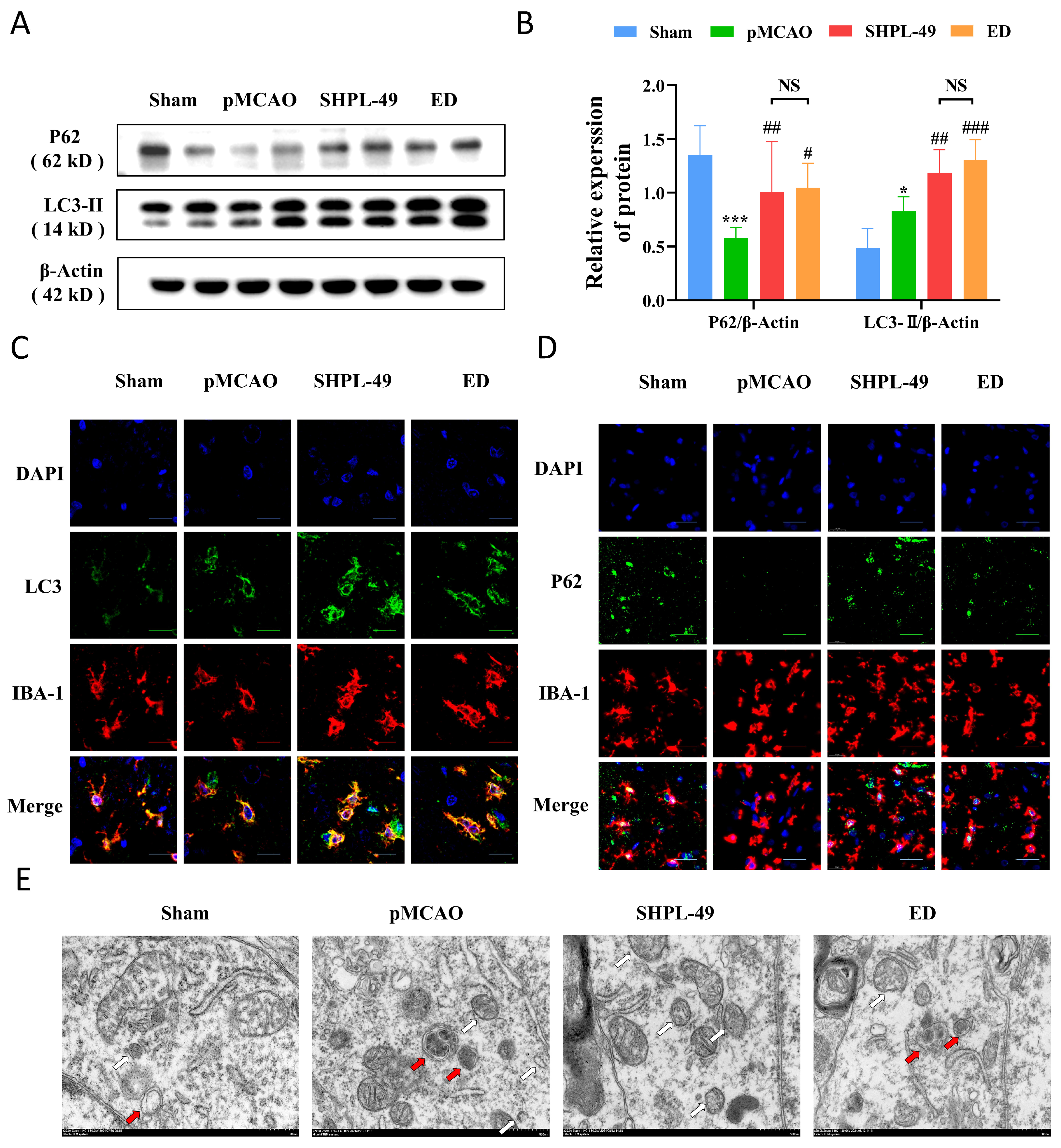
The protein P62 functions as an autophagic adapter and plays a crucial role in the degradation of proteins. During the process of autophagic lysosomal degradation, P62 binds to substrates and subsequently undergoes degradation by proteases. Consequently, an elevation in the level of P62 protein is commonly regarded as an indicator of suppressed autophagic activity [
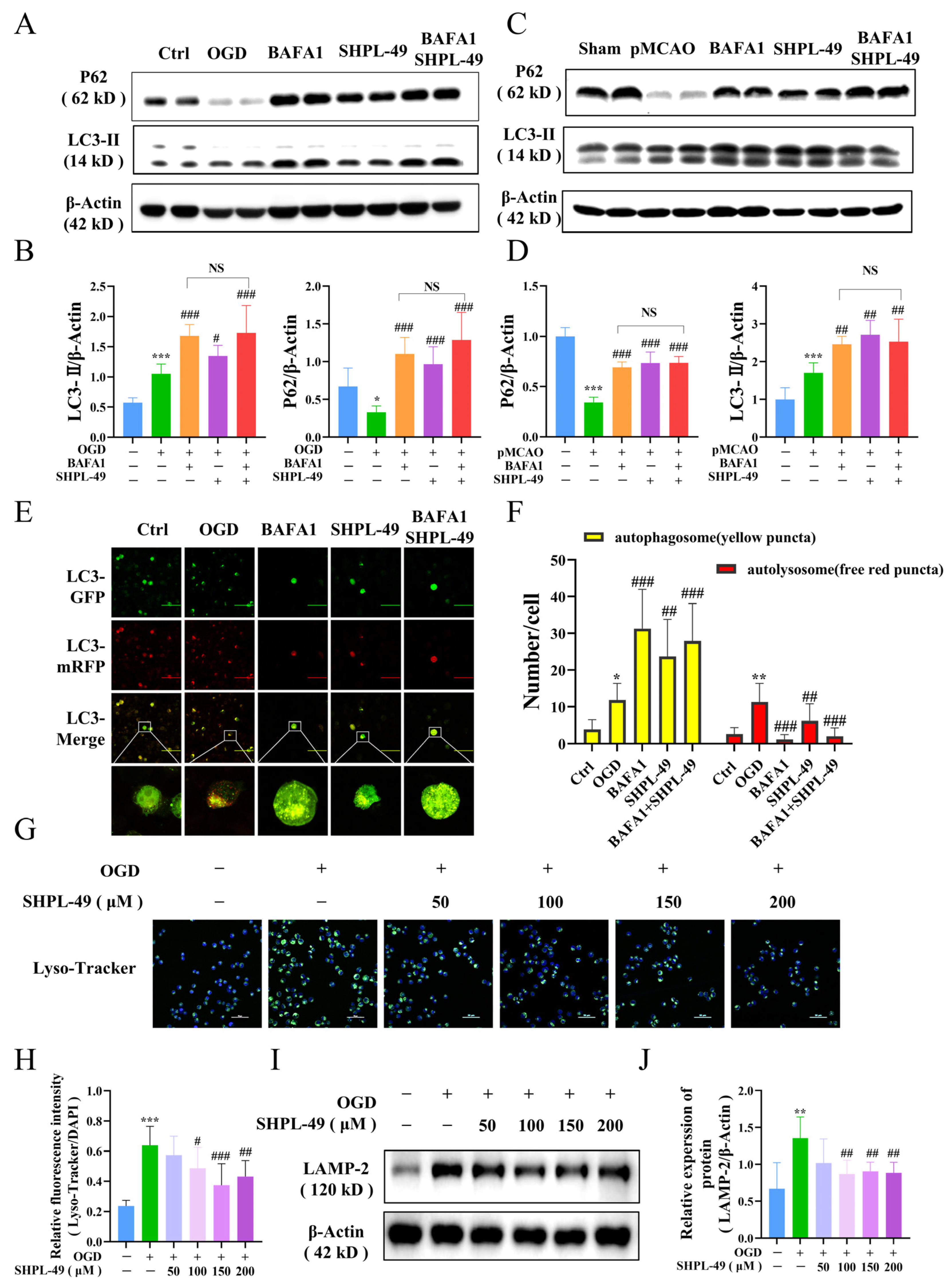
45]. To investigate the potential of SHPL-49 in modulating neuroinflammation through autophagy regulation, we examined its impact on cellular autophagic proteins and further assessed its effect in the presence of the autophagy inhibitor BAFA1. Our study revealed that treatment of OGD-BV2 cells with SHPL-49 resulted in elevated levels of LC3-II, P62, ATG5, and Beclin-1 proteins, potentially attributed to the inhibition of autophagic body degradation and subsequent protein accumulation. Subsequently, we conducted a further analysis of autophagy by introducing the autophagy inhibitor BAFA1. The fusion of autophagic bodies with lysosomes was inhibited by BAFA1. However, the co-administration of BAFA1 and SHPL-49 did not significantly alter the expression of LC3-II and P62 proteins compared to the BAFA1 group, thereby further confirming the inhibitory effects of SHPL-49 on autophagy. The inhibitory effects of SHPL-49 on lysosome activity were subsequently confirmed through the detection of lysosomal-associated membrane protein 2 (LAMP-2) and the visualization of lysosomes using Lyso-Tracker fluorescent dye, thereby providing further evidence for the inhibition of autophagic body fusion with lysosomes by SHPL-49.
The mounting evidence suggests an intricate network connection between autophagy and NF-κB, with the influence between them remaining contentious and contingent upon cell type and stimuli. The study conducted by Mingxia Zhou et al. [
46] used in vitro experiments to demonstrate that lipopolysaccharide (LPS) suppresses autophagy in colitis through the TLR4-MyD88-MAPK pathway, thereby orchestrating downstream NF-κB activation and resulting in the generation of proinflammatory cytokines and oxidative stress. Alfredo Criollo et al. [
47] reported that the activation of the NF-κB signaling pathway necessitates autophagic stimulation. Therefore, in order to investigate the involvement of autophagy in SHPL-49-mediated anti-inflammatory effects, an autophagy inducer, Rapa, was incorporated into the experiments. The findings demonstrated that the autophagy inducer Rapa significantly augmented NF-κB activation within the nucleus and enhanced the expression of inflammatory cytokines IL-6, IL-1β, and iNOS. However, the co-administration of SHPL-49 and Rapa effectively inhibits NF-κB activation and attenuates inflammatory response induced by Rapa. In conclusion, the inhibition of autophagy can reduce the inflammatory response triggered by ischemic stroke, while our compound SHPL-49 demonstrates its protective effects against ischemic stroke through microglial cell autophagy inhibition and the alleviation of neuroinflammation.
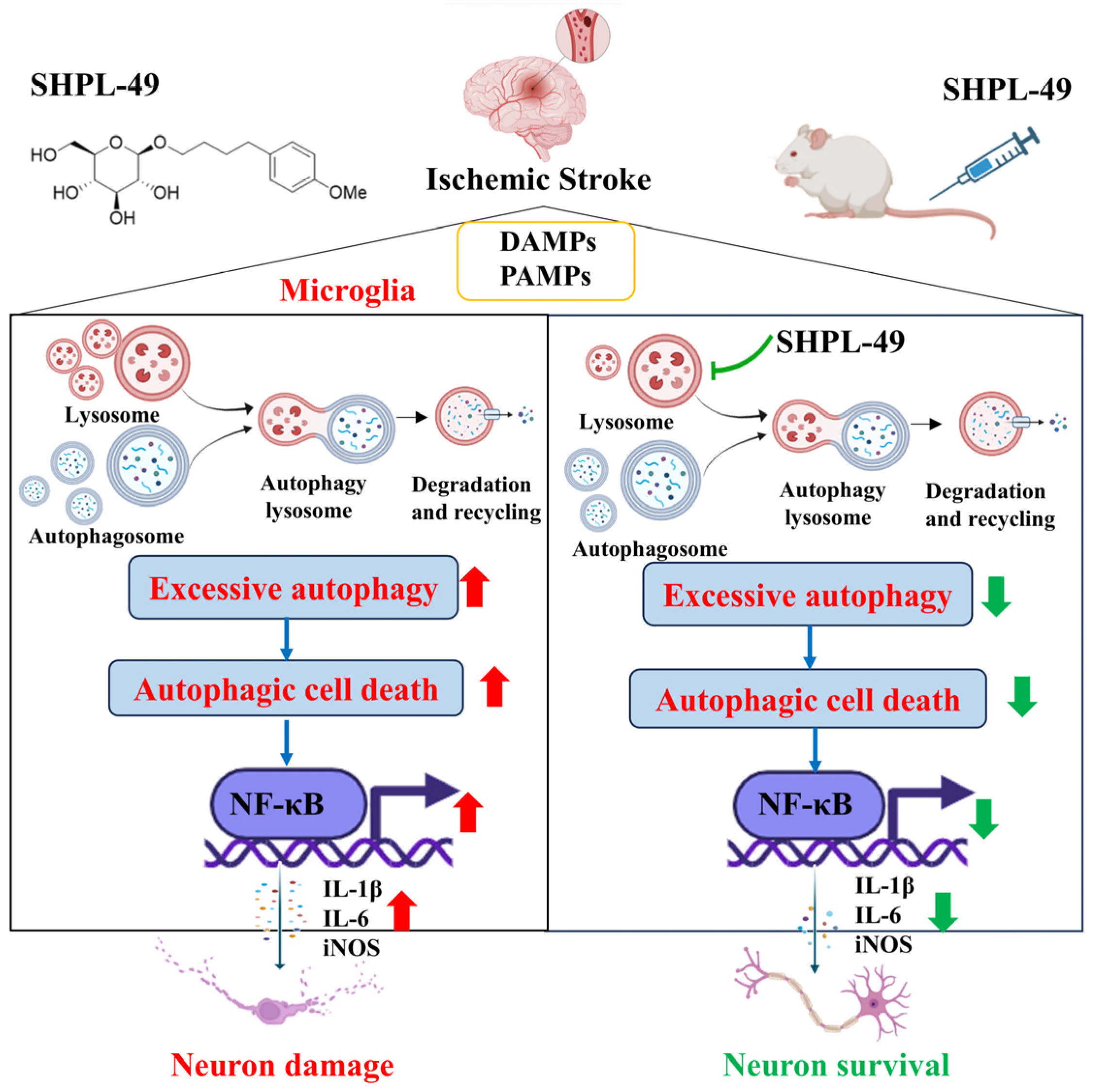
Based on previous pharmacological studies investigating the neuroprotective effects of SHPL-49 in rats with cerebral ischemia, we further explored the effects of SHPL-49 on microglial autophagy and neuroinflammation. Overall, as shown in the Schematic diagram (
Figure 6), the findings suggest that SHPL-49 hinders autophagy by suppressing lysosomal activity and the fusion between autophagosomes and lysosomes, thereby suppressing autophagy and reducing NF-κB pathway activation and exerting an anti-neuroinflammatory effect. This study provides valuable experimental data for researching treatments targeting neuroinflammation in ischemic stroke while also offering new potential active compounds for treating this condition and presenting a novel approach to developing and applying neuroprotective agents.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}