
Autophagy and Respiratory Viruses: Mechanisms, Viral Exploitation, and Therapeutic Insights
 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Influenza Virus and Autophagy
3. Respiratory Syncytial Virus and Autophagy
4. Coronaviruses and Autophagy
5. Human Parainfluenza Viruses and Autophagy
6. Adenovirus and Autophagy
7. Potential Therapeutic Approaches Using Autophagy
7.1. Current Clinical Trials Targeting Autophagy in Respiratory Virus Infections
7.2. Future Directions for Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACEIs | ACE inhibitors |
| AdV | Adenovirus |
| AMPK | AMP-activated protein kinase |
| Ang II | angiotensin II |
| ARDS | acute respiratory distress syndrome |
| ATG | autophagy-related gene |
| BAG3 | Bcl-2-associated athanogene 3 |
| CARD | caspase recruitment domain |
| CMA | chaperone-mediated autophagy |
| E1A | early region 1A |
| eNHE | endosomal Na+/H+ exchangers |
| F | fusion protein |
| Gal-8 | Galectin-8 |
| H | hemagglutinin-neuraminidase protein |
| HAdC-B7 | human adenovirus B7 |
| L | RNA polymerase |
| LC3 | microtubule-associated protein 1 light chain 3 beta |
| LRPPRC | leucine-rich pentatricopeptide repeat-containing protein |
| LRTI | lower respiratory tract infection |
| M | matrix protein |
| MAVS | mitochondrial antiviral signaling protein |
| MERS-CoV | Middle-East respiratory syndrome coronavirus |
| N | nucleocapsid protein |
| NCOA4 | nuclear receptor coactivator 4 |
| NSP | non-structural protein |
| P | phosphoprotein |
| PESP | PCBP1-AS1-encoded small protein |
| PVI | protein VI |
| QF | Qingfei |
| ROS | reactive oxygen species |
| RSV | respiratory syncytial virus |
| HPIV | Human parainfluenza virus |
| IAV | influenza A virus |
| IFN | interferon |
| IKK | NF-kB kinase |
| ISG15 | interferon-stimulated gene 15 |
| KPNA1 | karyopherin α1 |
| LDLR | low-density lipoprotein receptor |
| RIG-I | retinoic acid-inducible gene I |
| RLRs | retinoic acid-inducible gene I-like receptors |
| SKP2 | S-phase kinase-associated protein 2 |
| TRAF | TNF receptor-associated factor |
| TUFM | translation elongation factor mitochondrial |
| V-ATPase | vacuolar ATPase |
| VAMP8 | vesicle-associated membrane protein 8 |
| VPS34 | Class III Phosphatidylinositol 3-Kinase |
References
- Bender, R.G.; Sirota, S.B.; Swetschinski, L.R.; Dominguez, R.-M.V.; Novotney, A.; E Wool, E.; Ikuta, K.S.; Vongpradith, A.; Rogowski, E.L.B.; Doxey, M.; et al. Global, regional, and national incidence and mortality burden of non-COVID-19 lower respiratory infections and aetiologies, 1990–2021: A systematic analysis from the Global Burden of Disease Study 2021. Lancet Infect. Dis. 2024, 24, 974–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Shi, T.; Bont, L.J.; Chu, H.Y.; Zar, H.J.; Wahi-Singh, B.; Ma, Y.; Cong, B.; Sharland, E.; et al. Global disease burden of and risk factors for acute lower respiratory infections caused by respiratory syncytial virus in preterm infants and young children in 2019: A systematic review and meta-analysis of aggregated and individual participant data. Lancet 2024, 403, 1241–1253. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Shi, T.; Denouel, A.; Tietjen, A.K.; Campbell, I.; Moran, E.; Li, X.; Campbell, H.; Demont, C.; O Nyawanda, B.; Chu, H.Y.; et al. Global Disease Burden Estimates of Respiratory Syncytial Virus–Associated Acute Respiratory Infection in Older Adults in 2015: A Systematic Review and Meta-Analysis. J. Infect. Dis. 2019, 222, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Cilloniz, C.; Luna, C.M.; Hurtado, J.C.; Marcos, M.Á.; Torres, A. Respiratory viruses: Their importance and lessons learned from COVID-19. Eur. Respir. Rev. 2022, 31, 220051. [Google Scholar] [CrossRef]
- Shoar, S.; Musher, D.M. Etiology of community-acquired pneumonia in adults: A systematic review. Pneumonia 2020, 12, 1–10. [Google Scholar] [CrossRef]
- Burk, M.; El-Kersh, K.; Saad, M.; Wiemken, T.; Ramirez, J.; Cavallazzi, R. Viral infection in community-acquired pneumonia: A systematic review and meta-analysis. Eur. Respir. Rev. 2016, 25, 178–188. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Zhang, Y.-F.; Xu, Q.; Qiu, Y.; Lu, Q.-B.; Wang, T.; Zhang, X.-A.; Lin, S.-H.; Lv, C.-L.; Jiang, B.-G.; et al. Infection and co-infection patterns of community-acquired pneumonia in patients of different ages in China from 2009 to 2020: A national surveillance study. Lancet Microbe 2023, 4, 330–339. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, H.R.; Yu, H. Viral Respiratory Infections. In Hunter’s Tropical Medicine and Emerging Infectious Diseases, 10th ed.; Ryan, E.T., Hill, D.R., Solomon, T., Aronson, N.E., Endy, T.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 284–288. [Google Scholar]
- Chemaly, R.F.; Rathod, D.B.; Couch, R. Respiratory viruses. In Principle and Practice of Cancer Infectious Diseases; Safdar, A., Ed.; Springer Nature: London, UK, 2011; pp. 371–385. [Google Scholar]
- Chang, C.-C.; You, H.-L.; Huang, S.-T. Catechin inhibiting the H1N1 influenza virus associated with the regulation of autophagy. J. Chin. Med. Assoc. 2020, 83, 386–393. [Google Scholar] [CrossRef]
- Soudani, S.; Mafi, A.; Al Mayahi, Z.; Al Balushi, S.; Dbaibo, G.; Al Awaidy, S.; Amiche, A. A Systematic Review of Influenza Epidemiology and Surveillance in the Eastern Mediterranean and North African Region. Infect. Dis. Ther. 2022, 11, 15–52. [Google Scholar] [CrossRef]
- I Mazur, N.; Caballero, M.T.; Nunes, M.C. Severe respiratory syncytial virus infection in children: Burden, management, and emerging therapies. Lancet 2024, 404, 1143–1156. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mavunda, K.; Krilov, L.R. Current State of Respiratory Syncytial Virus Disease and Management. Infect. Dis. Ther. 2021, 10, 5–16. [Google Scholar] [CrossRef]
- Gebru, A.A.; Birhanu, T.; Wendimu, E.; Ayalew, A.F.; Mulat, S.; Abasimel, H.Z.; Kazemi, A.; Tadesse, B.A.; Deriba, B.S.; Zeleke, N.S.; et al. Global burden of COVID-19: Situational analyis and review. Hum. Antibodies 2021, 29, 139–148. [Google Scholar] [CrossRef]
- Heo, M.; Jeong, J.H.; Ju, S.; Lee, S.J.; Jeong, Y.Y.; Lee, J.D.; Yoo, J.W. Comparison of clinical features and outcomes between SARS-CoV-2 and non-SARS-CoV-2 respiratory viruses associated acute respiratory distress syndrome: Retrospective analysis. J. Clin. Med. 2022, 11, 2246. [Google Scholar] [CrossRef] [PubMed]
- Branche, A.R.; Falsey, A.R. Parainfluenza virus infection. Semin. Respir. Crit. Care. Med. 2016, 37, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Shieh, W.-J. Human adenovirus infections in pediatric population–An update on clinico-pathologic correlation. Biomed. J. 2021, 45, 38–49. [Google Scholar] [CrossRef]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Manzoor, R.; Igarashi, M.; Takada, A. Influenza A Virus M2 Protein: Roles from Ingress to Egress. Int. J. Mol. Sci. 2017, 18, 2649. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, T.; Hu, J.; Jin, S.; Wu, J.; Guan, X.; Wu, Y.; Cui, J. Targeting Selective Autophagy as a Therapeutic Strategy for Viral Infectious Diseases. Front. Microbiol. 2022, 13, 889835. [Google Scholar] [CrossRef]
- Shariq, M.; Malik, A.A.; Sheikh, J.A.; Hasnain, S.E.; Ehtesham, N.Z. Regulation of autophagy by SARS-CoV-2: The multifunctional contributions of ORF3a. J. Med. Virol. 2023, 95, e28959. [Google Scholar] [CrossRef]
- Chiok, K.; Pokharel, S.M.; Mohanty, I.; Miller, L.G.; Gao, S.-J.; Haas, A.L.; Tran, K.C.; Teng, M.N.; Bose, S. Human Respiratory Syncytial Virus NS2 Protein Induces Autophagy by Modulating Beclin1 Protein Stabilization and ISGylation. mBio 2022, 13, e0352821. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; Zeng, R.; Yang, J.; Liu, J.; Zhang, Z.; Song, X.; Yao, Z.; Ma, C.; Li, W.; et al. Respiratory Syncytial Virus Replication Is Promoted by Autophagy-Mediated Inhibition of Apoptosis. J. Virol. 2018, 92, e02193-17. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of Human Parainfluenza Virus Type 3 Blocks Autophagosome-Lysosome Fusion to Increase Virus Production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Piya, S.; White, E.J.; Klein, S.R.; Jiang, H.; McDonnell, T.J.; Gomez-Manzano, C.; Fueyo, J. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PLoS ONE 2011, 6, e29467. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the Therapeutic Potential of Delta-24-RGD in Brain Tumor Stem Cells: Role of Autophagic Cell Death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Zhang, W.; Dong, X.; Liu, M.; Chen, H.; Tang, B. The battle for autophagy between host and influenza A virus. Virulence 2021, 13, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Abdoli, A.; Alirezaei, M.; Mehrbod, P.; Forouzanfar, F. Autophagy: The multi-purpose bridge in viral infections and host cells. Rev. Med. Virol. 2018, 28, e1973. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Dastsooz, H.; Eshraghi, M.; Los, M.J.; Klionsky, D.J.; Ghavami, S. Autophagy: The Potential Link between SARS-CoV-2 and Cancer. Cancers 2021, 13, 5721. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, Z.; Castro-Gonzalez, S. Bidirectional interplay between SARS-CoV-2 and autophagy. mBio. 2023, 14, 01020–01023. [Google Scholar] [CrossRef]
- Iranpour, M.; Moghadam, A.R.; Yazdi, M.; Ande, S.R.; Alizadeh, J.; Wiechec, E.; Lindsay, R.; Drebot, M.; Coombs, K.M.; Ghavami, S. Apoptosis, autophagy and unfolded protein response pathways in Arbovirus replication and pathogenesis. Expert Rev. Mol. Med. 2016, 18, 1. [Google Scholar] [CrossRef] [PubMed]
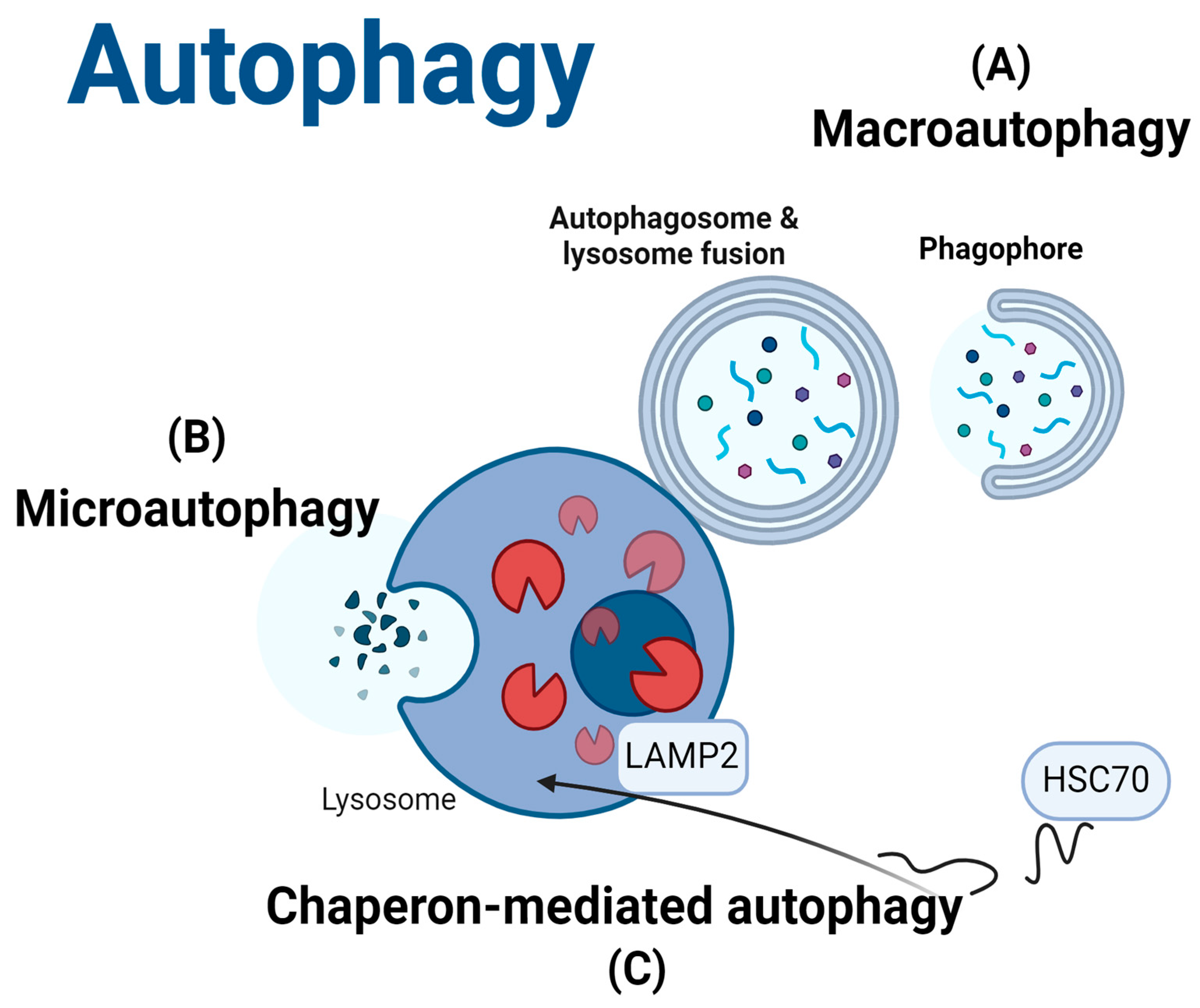
- Yang, Z.; Klionsky, D.J. An Overview of the Molecular Mechanism of Autophagy. Autophagy Infect. Immun. 2009, 335, 1–32. [Google Scholar] [CrossRef]
- Yamamoto, H.; Matsui, T. Molecular Mechanisms of Macroautophagy, Microautophagy, and Chaperone-Mediated Autophagy. J. Nippon. Med. Sch. 2024, 91, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Cordani, M.; Fiore, A.; Donadelli, M.; Gordon, J.W.; Klionsky, D.J.; Ghavami, S. The obesity-autophagy-cancer axis: Mechanistic insights and therapeutic perspectives. Semin. Cancer Biol. 2024, 99, 24–44. [Google Scholar] [CrossRef]
- Pirmoradi, L.; Shojaei, S.; Ghavami, S.; Zarepour, A.; Zarrabi, A. Autophagy and Biomaterials: A Brief Overview of the Impact of Autophagy in Biomaterial Applications. Pharmaceutics 2023, 15, 2284. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Rosa, S.C.d.S.; Weng, X.; Jacobs, J.; Lorzadeh, S.; Ravandi, A.; Vitorino, R.; Pecic, S.; Zivkovic, A.; Stark, H.; et al. Ceramides and ceramide synthases in cancer: Focus on apoptosis and autophagy. Eur. J. Cell Biol. 2023, 102, 151337. [Google Scholar] [CrossRef]
- Alizadeh, J.; Kavoosi, M.; Singh, N.; Lorzadeh, S.; Ravandi, A.; Kidane, B.; Ahmed, N.; Mraiche, F.; Mowat, M.R.; Ghavami, S. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers 2023, 15, 2195. [Google Scholar] [CrossRef]
- Hajiahmadi, S.; Lorzadeh, S.; Iranpour, R.; Karima, S.; Rajabibazl, M.; Shahsavari, Z.; Ghavami, S. Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy. Biology 2023, 12, 302. [Google Scholar] [CrossRef]
- Hayat, M.A. Overview of autophagy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–122. [Google Scholar]
- Dalvand, A.; da Silva Rosa, S.C.; Ghavami, S.; Marzban, H. Potential role of TGFBeta and autophagy in early crebellum development. Biochem. Biophys. Rep. 2022, 32, 101358. [Google Scholar]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. J. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Strippoli, R.; Trionfetti, F.; Behrooz, A.B.; Rumio, C.; Velasco, G.; Ghavami, S.; Marcucci, F. Immune checkpoints between epithelial-mesenchymal transition and autophagy: A conflicting triangle. Cancer Lett. 2024, 585, 216661. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Da Silva Rosa, S.C.; Martens, M.D.; Thliveris, J.; Ghavami, S.; Rampitsch, C.; Dolinsky, V.W.; Gordon, J.W.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2020, 17, 2257–2272. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Fayazbakhsh, F.; Mohammadi, M.; Babaei, Z.; Behrooz, A.B.; Tabasi, F.; Guan, T.; Beheshti, I.; Aghaei, M.; Klionsky, D.J.; et al. Sex and Region-Specific Disruption of Autophagy and Mitophagy in Alzheimer’s Disease: Linking Cellular Dysfunction to Cognitive Decline. bioRxiv 2024. Available online: https://pubmed.ncbi.nlm.nih.gov/39554142/ (accessed on 10 December 2024).
- Reggio, A.; Buonomo, V.; Grumati, P. Eating the unknown: Xenophagy and ER-phagy are cytoprotective defenses against pathogens. Exp. Cell Res. 2020, 396, 112276. [Google Scholar] [CrossRef]
- Al-Bari, A.A.; Ito, Y.; Ahmed, S.; Radwan, N.; Ahmed, H.S.; Eid, N. Targeting Autophagy with Natural Products as a Potential Therapeutic Approach for Cancer. Int. J. Mol. Sci. 2021, 22, 9807. [Google Scholar] [CrossRef]
- Kazyken, D.; Dame, S.G.; Wang, C.; Wadley, M.; Fingar, D.C. Unexpected roles for AMPK in the suppression of autophagy and the reactivation of MTORC1 signaling during prolonged amino acid deprivation. Autophagy 2024, 20, 2017–2040. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Klionsky, D.J. Highlights in the fight against COVID-19: Does autophagy play a role in SARS-CoV-2 infection? Autophagy 2020, 16, 2123–2127. [Google Scholar] [CrossRef]
- Siri, M.; Dastghaib, S.; Zamani, M.; Rahmani-Kukia, N.; Geraylow, K.R.; Fakher, S.; Keshvarzi, F.; Mehrbod, P.; Ahmadi, M.; Mokarram, P.; et al. Autophagy, Unfolded Protein Response, and Neuropilin-1 Cross-Talk in SARS-CoV-2 Infection: What Can Be Learned from Other Coronaviruses. Int. J. Mol. Sci. 2021, 22, 5992. [Google Scholar] [CrossRef]
- Yeganeh, B.; Rezaei, M.A.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi MGeramizadeh, B.; Samali, A.; Bagheri, L.K.; Post, M.; Peymani, P.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol 2015, 21, 13225–13239. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ling, J.; Li, J. Is Autophagy a Friend or Foe in SARS-CoV-2 Infection? Viruses 2024, 16, 1491. [Google Scholar] [CrossRef]
- Uyeki, T.M.; Hui, D.S.; Zambon, M.; Wentworth, D.E.; Monto, A.S. Influenza. Lancet. 2022, 400, 693–706. [Google Scholar] [CrossRef] [PubMed]
- CDC. Seasonal Influenza–Annual Epidemiological Report for 2022/2023, 2023. European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en/publications-data/seasonal-influenza-annual-epidemiological-report-20222023 (accessed on 10 December 2024).
- CDC. 2023–2024 U.S. Flu Season: Preliminary In-season Burden Estimates, 2024 European Centre for Disease Prevention and Control. Available online: https://www.bing.com/ck/a?!&&p=8fccedab4df6d46795c2de0817885d894e53c09975ff01cd4367621c713313cdJmltdHM9MTc0MTIxOTIwMA&ptn=3&ver=2&hsh=4&fclid=26f551ea-06b7-6b4a-1b88-44b907aa6a43&psq=2023-2024+U.S.+Flu+Season%3a+Preliminary+in-season+burden+estimates.+2024+%5bcited+2024+21+July%5d%3b+&u=a1aHR0cHM6Ly9zdGFja3MuY2RjLmdvdi92aWV3L2NkYy8xNTY5NTkvY2RjXzE1Njk1OV9EUzEucGRm&ntb=1 (accessed on 10 December 2024).
- Pumarola, T.; Diez-Domingo, J.; Martinon-Torres, F.; Redondo, M.E.; de Lejarazu, L.R.O.; Carmo, M.; Bizouard, G.; Drago, G.; Lopez-Belmonte, J.; L’ Bricout, H.; et al. Excess Hospitalizations and Mortality Associated with Seasonal Influenza in Spain, 2008–2018. BMC Infect. Dis. 2023, 23, 86. [Google Scholar] [CrossRef]
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A brief review of influenza virus infection. J. Med. Virol. 2021, 93, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Ouranos, K.; Vassilopoulos, S.; Vassilopoulos, A.; Shehadeh, F.; Mylonakis, E. Cumulative incidence and mortality rate of cardiovascular complications due to laboratory-confirmed influenza virus infection: A systematic review and meta-analysis. Rev. Med. Virol. 2023, 34, e2497. [Google Scholar] [CrossRef]
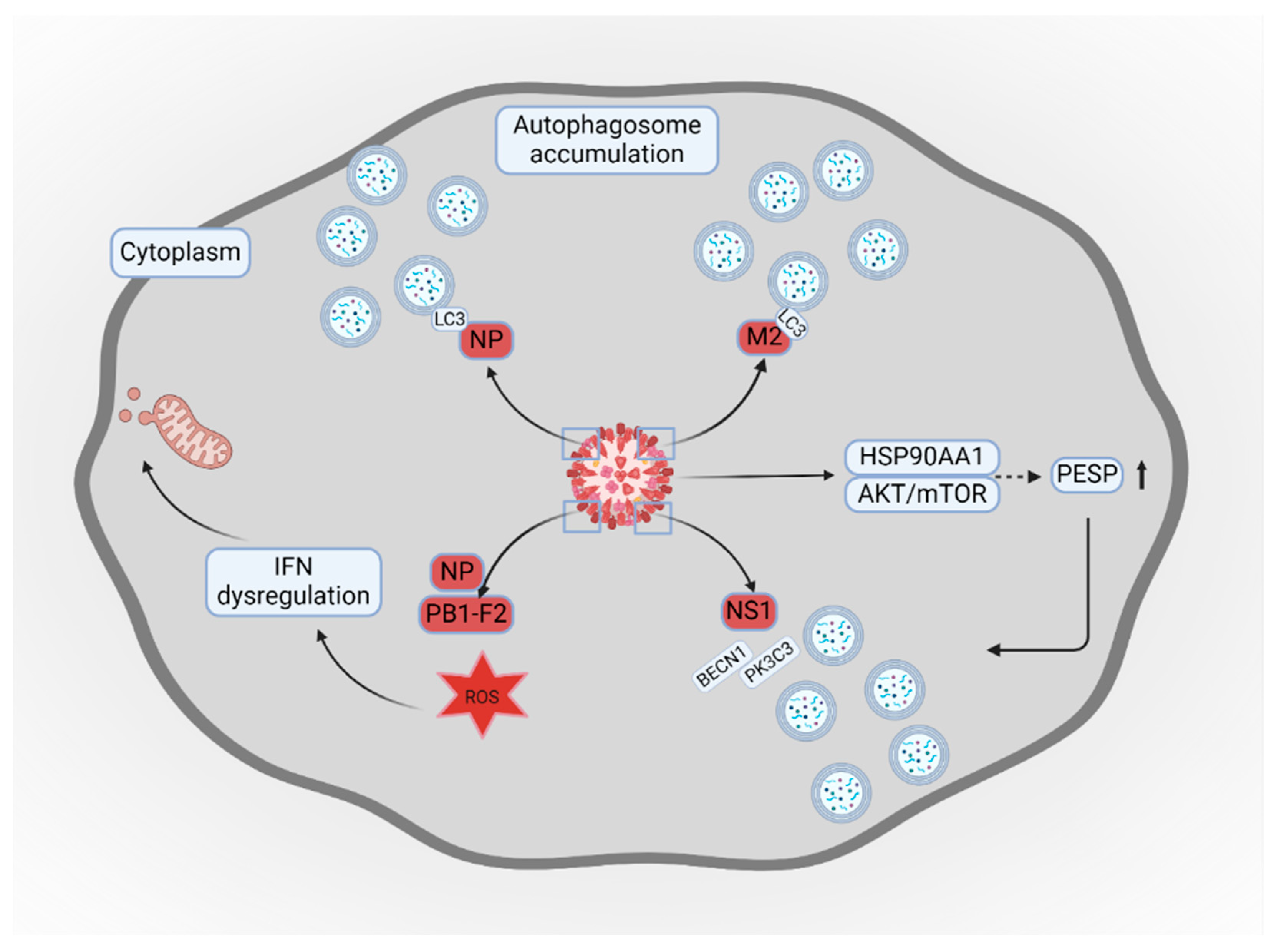
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Z.; Lin, C.; Ren, H.; Li, Y.; Zhang, Y.; Qu, Y.; Li, H.; Ma, S.; Xia, H.; et al. A/(H1N1) pdm09 NS1 promotes viral replication by enhancing autophagy through hijacking the IAV negative regulatory factor LRPPRC. Autophagy 2022, 19, 1533–1550. [Google Scholar] [CrossRef]
- Guo, R.; Liu, H.; Su, R.; Mao, Q.; Zhao, M.; Zhang, H.; Mu, J.; Zhao, N.; Wang, Y.; Hao, Y. Tanreqing injection inhibits influenza virus replication by promoting the fusion of autophagosomes with lysosomes: An integrated pharmacological study. J. Ethnopharmacol. 2024, 331, 118159. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, H.; Li, C. Advances in deciphering the interactions between viral proteins of influenza A virus and host cellular proteins. Cell Insight 2023, 2, 100079. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 Phosphorylation of p62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, T.; Lin, L.; Zhang, Y.; Peng, X.; Yan, Y.; Lei, J.; Zhou, J.; Hu, B. Influenza A Virus Induces Autophagy by Its Hemagglutinin Binding to Cell Surface Heat Shock Protein 90AA1. Front. Microbiol. 2020, 11, 566348. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Huang, G.; Wang, L.; Zhang, X.; Liu, J.; Yin, Z.; Guo, G.; Chen, Y.; Wang, S.; Chen, J.-L. A small protein encoded by PCBP1-AS1 is identified as a key regulator of influenza virus replication via enhancing autophagy. PLoS Pathog. 2024, 20, e1012461. [Google Scholar] [CrossRef]
- Zou, J.; Yue, F.; Li, W.; Song, K.; Jiang, X.; Yi, J.; Liu, L. Autophagy Inhibitor LRPPRC Suppresses Mitophagy through Interaction with Mitophagy Initiator Parkin. PLoS ONE 2014, 9, e94903. [Google Scholar] [CrossRef]
- Zou, J.; Yue, F.; Jiang, X.; Li, W.; Yi, J.; Liu, L. Mitochondrion-associated protein LRPPRC suppresses the initiation of basal levels of autophagy via enhancing Bcl-2 stability. Biochem. J. 2013, 454, 447–457. [Google Scholar] [CrossRef]
- Zhang, R.-H.; Zhang, H.-L.; Li, P.-Y.; Li, C.-H.; Gao, J.-P.; Li, J.; Xu, T.; Wang, X.-J.; Wang, C.-L.; Zhang, H.-C.; et al. Autophagy is involved in the replication of H9N2 influenza virus via the regulation of oxidative stress in alveolar epithelial cells. Virol. J. 2021, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juarbe, N.; Riegler, A.N.; Jureka, A.S.; Gilley, R.P.; Brand, J.D.; Trombley, J.E.; Scott, N.R.; Platt, M.P.; Dube, P.H.; Petit, C.M.; et al. Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin-Mediated Necroptosis. Cell Rep. 2020, 32, 108062. [Google Scholar] [CrossRef]
- Dalskov, L.; Gad, H.H.; Hartmann, R. Viral recognition and the antiviral interferon response. EMBO J. 2023, 42, e112907. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2020, 17, 496–511. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, S.; Liu, M.; Wei, Y.; Wang, Q.; Shen, W.; Lei, C.-Q.; Zhu, Q. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 2023, 19, 1916–1933. [Google Scholar] [CrossRef]
- Xu, S.; Han, L.; Wei, Y.; Zhang, B.; Wang, Q.; Liu, J.; Liu, M.; Chen, Z.; Wang, Z.; Chen, H.; et al. MicroRNA-200c-targeted contactin 1 facilitates the replication of influenza A virus by accelerating the degradation of MAVS. PLoS Pathog. 2022, 18, e1010299. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Xu, W.; Ai, X.; Zhu, Y.; Geng, P.; Niu, Y.; Zhu, H.; Zhou, W.; Huang, H.; Shi, X. Autophagy and Exosome Coordinately Enhance Macrophage M1 Polarization and Recruitment in Influenza A Virus Infection. Front. Immunol. 2022, 13, 722053. [Google Scholar] [CrossRef]
- Cole, S.L.; Dunning, J.; Kok, W.L.; Benam, K.H.; Benlahrech, A.; Repapi, E.; Martinez, F.O.; Drumright, L.; Powell, T.J.; Bennett, M.; et al. M1-like monocytes are a major immunological determinant of severity in previously healthy adults with life-threatening influenza. J. Clin. Investig. 2017, 2, e91868. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Drexler, I. Targeting Autophagy in Innate Immune Cells: Angel or Demon During Infection and Vaccination? Front Immunol. 2020, 11, 460. [Google Scholar] [CrossRef]
- Zang, F.; Chen, Y.; Lin, Z.; Cai, Z.; Yu, L.; Xu, F.; Wang, J.; Zhu, W.; Lu, H. Autophagy is involved in regulating the immune response of dendritic cells to influenza A (H1N1) pdm09 infection. Immunology 2016, 148, 56–69. [Google Scholar] [CrossRef]
- Puleston, D.J.; Simon, A.K. Autophagy in the immune system. Immunology 2013, 141, 1–8. [Google Scholar] [CrossRef]
- Liu, S.; Mok, B.W.-Y.; Deng, S.; Liu, H.; Wang, P.; Song, W.; Chen, P.; Huang, X.; Zheng, M.; Lau, S.-Y.; et al. Mammalian cells use the autophagy process to restrict avian influenza virus replication. Cell Rep. 2021, 35, 109213. [Google Scholar] [CrossRef]
- Chang, C.; You, H.; Su, H.; Hung, I.; Kao, C.; Huang, S. Anti-influenza A (H1N1) virus effect of gallic acid through inhibition of virulent protein production and association with autophagy. Food Sci. Nutr. 2023, 12, 1605–1615. [Google Scholar] [CrossRef]
- Godbole, N.M.; Sinha, R.A.; Tiwari, S.; Pawar, S.D.; Dhole, T. Analysis of influenza virus-induced perturbation in autophagic flux and its modulation during Vitamin D3 mediated anti-apoptotic signaling. Virus Res. 2020, 282, 197936. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, J.; Shan, C.; Song, X.; Yang, J.; Xu, H.; Ou, D. Baicalin reduced injury of and autophagy-related gene expression in RAW264.7 cells infected with H6N6 avian influenza virus. Heliyon 2024, 10, e32645. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, C.-L.; Yan, Y.-L.; Zhang, F.-L.; Chen, J.; Hu, Z.-Y.; He, J.; Meng, X.-Y.; Wu, Q.-F. Inhibitory Effects and Related Molecular Mechanisms of Huanglian-Ganjiang Combination Against H1N1 Influenza Virus. Rev. Bras. Farmacogn. 2023, 33, 514–522. [Google Scholar] [CrossRef]
- Zhang, R.; Chi, X.; Wang, S.; Qi, B.; Yu, X.; Chen, J.-L. The Regulation of Autophagy by Influenza A Virus. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Choi, J.-G.; Lee, H.; Kim, Y.S.; Hwang, Y.-H.; Oh, Y.-C.; Lee, B.; Moon, K.M.; Cho, W.-K.; Ma, J.Y. Aloe vera and its Components Inhibit Influenza A Virus-Induced Autophagy and Replication. Am. J. Chin. Med. 2019, 47, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013, 4, e562. [Google Scholar] [CrossRef]
- Mazel-Sanchez, B.; Iwaszkiewicz, J.; Bonifacio, J.P.P.; Silva, F.; Niu, C.; Strohmeier, S.; Eletto, D.; Krammer, F.; Tan, G.; Zoete, V.; et al. Influenza A Viruses Balance ER Stress with Host Protein Synthesis Shutoff. Proc. Natl. Acad. Sci. USA 2021, 118, e2024681118. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Rahim, M.; Klonisch, T.; Halayko, A.; Coombs, K. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta–Mol. Cell Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Bächi, T.; Howe, C. Morphogenesis and Ultrastructure of Respiratory Syncytial Virus. J. Virol. 1973, 12, 1173–1180. [Google Scholar] [CrossRef]
- Bourgeois, C.; Bour, J.B.; Lidholt, K.; Gauthray, C.; Pothier, P. Heparin-Like Structures on Respiratory Syncytial Virus Are Involved in Its Infectivity In Vitro. J. Virol. 1998, 72, 7221–7227. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S.; Boyapalle, S. Epidemiologic, Experimental, and Clinical Links between Respiratory Syncytial Virus Infection and Asthma. Clin. Microbiol. Rev. 2008, 21, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Blanken, M.O.; Rovers, M.M.; Molenaar, J.M.; Winkler-Seinstra, P.L.; Meijer, A.; Kimpen, J.L.; Bont, L. Respiratory Syncytial Virus and Recurrent Wheeze in Healthy Preterm Infants. N. Engl. J. Med. 2013, 368, 1791–1799. [Google Scholar] [CrossRef]
- Griffiths, C.D.; Bilawchuk, L.M.; McDonough, J.E.; Jamieson, K.C.; Elawar, F.; Cen, Y.; Duan, W.; Lin, C.; Song, H.; Casanova, J.L.; et al. IGF1R is an Entry Receptor for Respiratory Syncytial Virus. Nature 2020, 583, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Kieser, Q.J.; Granoski, M.J.; McClelland, R.D.; Griffiths, C.; Bilawchuk, L.M.; Stojic, A.; Elawar, F.; Jamieson, K.; Proud, D.; Marchant, D.J. Actin cytoskeleton remodeling disrupts physical barriers to infection and presents entry receptors to respiratory syncytial virus. J. Gen. Virol. 2023, 104, 001923. [Google Scholar] [CrossRef]
- Song, Q.; Zhu, H.; Qiu, M.; Cai, J.; Hu, Y.; Yang, H.; Rao, S.; Li, Y.; Li, M.; Hu, L.; et al. A new mechanism of respiratory syncytial virus entry inhibition by small-molecule to overcome K394R-associated resistance. mBio 2024, 15, e0138524. [Google Scholar] [CrossRef]
- Wilson, E.; Goswami, J.; Baqui, A.H.; Doreski, P.A.; Perez-Marc, G.; Zaman, K.; Monroy, J.; Duncan, C.J.; Ujiie, M.; Rämet, M.; et al. Efficacy and Safety of an mRNA-Based RSV PreF Vaccine in Older Adults. N. Engl. J. Med. 2023, 389, 2233–2244. [Google Scholar] [CrossRef]
- Jastorff, A.; Gymnopoulou, E.; Salas, J.; Merrall, E.; Buntinx, E.; Martin, C.; Askling, H.H.; Schenkenberger, I.; Yuste, A.C.; Smith, W.; et al. Safety and Immunogenicity of the Ad26/protein preF RSV Vaccine in Adults Aged 18 to 59 Years with and Without At-Risk Comorbidities for Severe Respiratory Syncytial Virus Disease: A Phase 3, Randomized, Controlled, Immunobridging Trial. Vaccine 2025, 43, 126514. [Google Scholar] [CrossRef]
- Bramante, C.T.; Beckman, K.B.; Mehta, T.; Karger, A.B.; Odde, D.J.; Tignanelli, C.J.; Buse, J.B.; Johnson, D.M.; Watson, R.H.B.; Daniel, J.J.; et al. Metformin Reduces SARS-CoV-2 in a Phase 3 Randomized Placebo Controlled Clinical Trial. medRxiv 2023. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC10275003/ (accessed on 10 December 2024).
- Huang, L.; Schibler, A.; Huang, Y.; Tai, A.; Chi, H.; Chieng, C.; Wang, J.; Goldbart, A.; Tang, S.; Huang, Y.; et al. Safety and efficacy of AK0529 in respiratory syncytial virus-infected infant patients: A phase 2 proof-of-concept trial. Influ. Other Respir. Viruses 2023, 17, e13176. [Google Scholar] [CrossRef]
- Han, B.; Wang, Y.; Zheng, M. Inhibition of Autophagy Promotes Human RSV NS1-induced Inflammation and Apoptosis. Exp. Ther. Med. 2021, 22, 1054. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wu, B.; Wang, Y.; He, H.; Lin, Z.; Tan, J.; Yang, L.; Kamp, D.W.; Zhou, X.; Tang, J.; et al. Particulate matter 2.5 induces autophagy via inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin kinase signaling pathway in human bronchial epithelial cells. Mol. Med. Rep. 2015, 12, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Azman, A.F.; Chia, S.L.; Sekawi, Z.; Yusoff, K.; Ismail, S. Inhibition of Autophagy Does Not Affect Innate Cytokine Production in Human Lung Epithelial Cells During Respiratory Syncytial Virus Infection. Viral Immunol. 2021, 34, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; Vergara, H.S.J.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 2005, 11, 56–62. [Google Scholar] [CrossRef]
- Yu, L.; Wang, J.; Zou, Y.; Zeng, H.; Cheng, W.; Jing, X. Qingfei oral liquid inhibited autophagy to alleviate inflammation via mTOR signaling pathway in RSV-infected asthmatic mice. Biomed. Pharmacother. 2021, 138, 111449. [Google Scholar] [CrossRef]
- Lin, L.; An, L.; Chen, H.; Feng, L.; Lu, M.; Liu, Y.; Chu, C.; Shan, J.; Xie, T.; Wang, X.; et al. Integrated Network Pharmacology and Lipidomics to Reveal the Inhibitory Effect of Qingfei Oral Liquid on Excessive Autophagy in RSV-Induced Lung Inflammation. Front. Pharmacol. 2021, 12, 777689. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Baker, H.; Vézina, C. Rapamycin (AY-22,989), a New Antifungal Antibiotic. II. Fermentation, Isolation and Characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.-S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Huckestein, B.R.; Zeng, K.; Westcott, R.; Alder, J.K.; Antos, D.; Kolls, J.K.; Alcorn, J.F. Mammalian Target of Rapamycin Complex 1 Activation in Macrophages Contributes to Persistent Lung Inflammation following Respiratory Tract Viral Infection. Am. J. Pathol. 2023, 194, 384–401. [Google Scholar] [CrossRef]
- Husain, A.; Byrareddy, S.N. Rapamycin as a potential repurpose drug candidate for the treatment of COVID-19. Chem. Interact. 2020, 331, 109282. [Google Scholar] [CrossRef]
- de Freitas, D.D.N.; Gassen, R.B.; Fazolo, T.; de Souza, A.P.D. Rapamycin increases RSV RNA levels and survival of RSV-infected dendritic cell depending on T cell contact. Toxicol. Vitr. 2016, 36, 114–119. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Sirviö, V.A.; Agip, A.-N.A.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ray, A.; Sadasivam, B. Metformin in COVID-19: A possible role beyond diabetes. Diabetes Res. Clin. Pract. 2020, 164, 108183. [Google Scholar] [CrossRef]
- Esam, Z. A proposed mechanism for the possible therapeutic potential of Metformin in COVID-19. Diabetes Res. Clin. Pract. 2020, 167, 108282. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H+-ATPase (V-ATPase): From proton pump to signaling complex in health and disease. Am. J. Physiol. Physiol. 2021, 320, 92-C414. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, H.; Tandel, D.; Siddiqui, A.H.; Harshan, K.H. Metformin Suppresses SARS-CoV-2 in Cell Culture. Virus Res. 2023, 323, 199010. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Xu, W.; Chen, J.; Tang, Y.; Xiong, S.; Li, Y.; Zhang, H.; Li, M.; Liu, Z. Cholesterol-rich lysosomes induced by respiratory syncytial virus promote viral replication by blocking autophagy flux. Nat. Commun. 2024, 15, 6311. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, W.; Liu, X.; Bai, J.; Jia, Q.; Shi, Q. Procyanidin A1 alleviates DSS-induced ulcerative colitis via regulating AMPK/mTOR/p70S6K-mediated autophagy. J. Physiol. Biochem. 2022, 78, 213–227. [Google Scholar] [CrossRef]
- Sun, D.; Tao, W.; Zhang, F.; Shen, W.; Tan, J.; Li, L.; Meng, Q.; Chen, Y.; Yang, Y.; Cheng, H. Trifolirhizin induces autophagy-dependent apoptosis in colon cancer via AMPK/mTOR signaling. Signal Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wu, W.; Xiong, Z.; Yu, X.; Ye, Z.; Wu, Z. Targeting autophagy drug discovery: Targets, indications and development trends. Eur. J. Med. Chem. 2024, 267, 116117. [Google Scholar] [CrossRef] [PubMed]
- Wildenbeest, J.G.; Lowe, D.M.; Standing, J.F.; Butler, C.C. Respiratory syncytial virus infections in adults: A narrative review. Lancet Respir. Med. 2024, 12, 822–836. [Google Scholar] [CrossRef]
- Alsaleh, G.; Panse, I.; Swadling, L.; Zhang, H.; Richter, F.; Meyer, A.; Lord, J.; Barnes, E.; Klenerman, P.; Green, C.; et al. Autophagy in T cells from aged donors is maintained by spermidine, and correlates with function and vaccine responses. eLife 2020, 9, e57950. [Google Scholar] [CrossRef]
- Merkley, S.D.; Chock, C.J.; Yang, X.O.; Harris, J.; Castillo, E.F. Modulating T Cell Responses via Autophagy: The Intrinsic Influence Controlling the Function of Both Antigen-Presenting Cells and T Cells. Front. Immunol. 2018, 9, 2914. [Google Scholar] [CrossRef] [PubMed]
- Faramarzi, A.; Norouzi, S.; Dehdarirad, H.; Aghlmand, S.; Yusefzadeh, H.; Javan-Noughabi, J. The global economic burden of COVID-19 disease: A comprehensive systematic review and meta-analysis. Syst. Rev. 2024, 13, 174. [Google Scholar] [CrossRef]
- Grellet, E.; L’Hôte, I.; Goulet, A.; Imbert, I. Replication of the coronavirus genome: A paradox among positive-strand RNA viruses. J. Biol. Chem. 2022, 298, 101923. [Google Scholar] [CrossRef]
- Zappulli, V.; Ferro, S.; Bonsembiante, F.; Brocca, G.; Calore, A.; Cavicchioli, L.; Centelleghe, C.; Corazzola, G.; De Vreese, S.; Gelain, M.E.; et al. Pathology of Coronavirus Infections: A Review of Lesions in Animals in the One-Health Perspective. Animals 2020, 10, 2377. [Google Scholar] [CrossRef]
- Luo, X.; Zhou, G.-Z.; Zhang, Y.; Peng, L.-H.; Zou, L.-P.; Yang, Y.-S. Coronaviruses and Gastrointestinal Diseases. Mil. Med. Res. 2020, 7, 49. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A. Angiotensin-Converting Enzyme 2 (ACE2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. Histochem. J. 2020, 51, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, Y.; Zeng, Z.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-Converting Enzyme 2/angiotensin-(1–7)/Mas Axis Prevents Lipopolysaccharide–Induced Apoptosis of Pulmonary Microvascular Endothelial Cells by Inhibiting JNK/NF–κB Pathways. Sci. Rep. 2015, 5, 8209. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
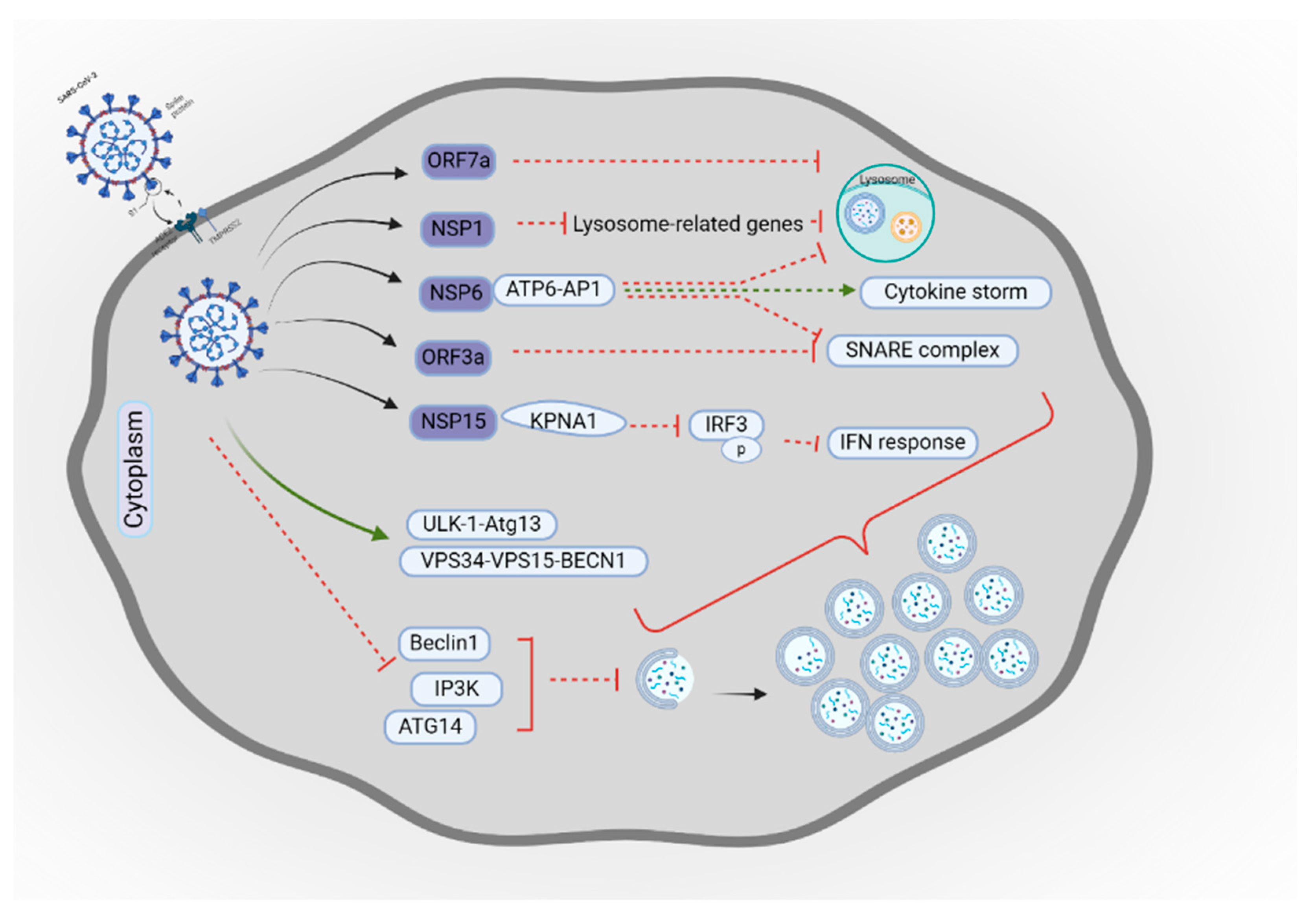
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated Dysregulation of Metabolism and Autophagy Uncovers Host-Targeting Antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Sadeghdoust, M.; Aligolighasemabadi, F.; Dehesh, T.; Taefehshokr, N.; Sadeghdoust, A.; Kotfis, K.; Hashemiattar, A.; Ravandi, A.; Aligolighasemabadi, N.; Vakili, O.; et al. The Effects of Statins on Respiratory Symptoms and Pulmonary Fibrosis in COVID-19 Patients with Diabetes Mellitus: A Longitudinal Multicenter Study. Arch. Immunol. Ther. Exp. 2023, 71, 8. [Google Scholar] [CrossRef]
- Wang, A.-M.; Gao, S.; Zhang, Z.-M.; Shen, Z.-L.; Gao, K.; Chang, L.; Guo, Y.; Li, Z.; Wang, W. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen. Res. 2016, 11, 977–982. [Google Scholar] [CrossRef]
- Han, F.; Xiao, Q.; Peng, S.; Che, X.; Jiang, L.; Shao, Q.; He, B. Atorvastatin ameliorates LPS-induced inflammatory response by autophagy via AKT/mTOR signaling pathway. J. Cell. Biochem. 2017, 119, 1604–1615. [Google Scholar] [CrossRef]
- Peymani, P.; Dehesh, T.; Aligolighasemabadi, F.; Sadeghdoust, M.; Kotfis, K.; Ahmadi, M.; Mehrbod, P.; Iranpour, P.; Dastghaib, S.; Nasimian, A.; et al. Statins in patients with COVID-19: A retrospective cohort study in Iranian COVID-19 patients. Transl. Med. Commun. 2021, 6, 3. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, Y.; Yang, X.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, Z.; et al. ORF3a Mediated-Incomplete Autophagy Facilitates SARS-CoV-2 Replication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Bozzo, C.P.; Aftab, W. Systematic Functional Analysis of SARS-CoV-2 Proteins Uncovers Viral Innate Immune Antagonists and Remaining Vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 Virus SARS-CoV-2 Blocks HOPS Complex-Mediated Assembly of the SNARE Complex Required for Autolysosome Formation. Dev. Cell 2021, 56, 427–442. [Google Scholar] [CrossRef]
- Koepke, L.; Hirschenberger, M.; Hayn, M.; Kirchhoff, F.; Sparrer, K.M. Manipulation of Autophagy by SARS-CoV-2 Proteins. Autophagy 2021, 17, 2659–2661. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X. SARS-CoV-2 Non-Structural Protein 6 Triggers NLRP3-dependent Pyroptosis by Targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254. [Google Scholar] [CrossRef]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef]
- Hulseberg, C.E.; Fénéant, L.; Szyman’ska, K.M.; Szyman’ska-De Wijs, S.; Kessler, N.P.; Nelson, E.A.; Shoemaker, C.J.; Schmaljohn, C.S.; Polyak, S.J.; White, J.M.; et al. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses Downloaded from. J. Virol. 2019, 93, e02185-18. [Google Scholar] [CrossRef]
- Kumar, S.; Javed, R.; Mudd, M.; Pallikkuth, S.; Lidke, K.A.; Jain, A.; Tangavelou, K.; Gudmundsson, S.R.; Ye, C.; Rusten, T.E.; et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 2021, 184, 5950–5969. [Google Scholar] [CrossRef]
- Feng, Y.; Pan, Z.; Wang, Z.; Lei, Z.; Yang, S.; Zhao, H.; Wang, X.; Yu, Y.; Han, Q.; Zhang, J. MERS-CoV nsp1 Regulates Autophagic Flux via mTOR Signalling and Dysfunctional Lysosomes. Emerg. Microbes. Infect. 2022, 11, 2529–2543. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The interplay between emerging human coronavirus infections and autophagy. Emerg. Microbes Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.-C.; Pan, B.-S.; Manne, R.; Li, H.-Y.; Lin, H.-K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.d.S.; Leao, A.H.F.F.; Erustes, A.G.; Morais, I.B.d.M.; Vrechi, T.A.d.M.; Zamarioli, L.d.S.; Pereira, C.A.S.; Marchioro, L.d.O.; Sperandio, L.P.L.; Isis, V.F. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 4067. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a Less Toxic Derivative of Chloroquine, is Effective in Inhibiting SARS-CoV-2 Infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Le Bideau, M.; Duflot, I.; Jardot, P.; Rolland, C.; Boxberger, M.; Wurtz, N.; Rolain, J.M.; Colson, P.; La Scola, B. In vitro Testing of Combined Hydroxychloroquine and Azithromycin on SARS-CoV-2 Shows Synergistic Effect. Microb. Patog. 2020, 145, 104228. [Google Scholar] [CrossRef]
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine does not Inhibit Infection of Human Lung Cells with SARS-CoV-2. Nature 2020, 585, 588–590. [Google Scholar] [CrossRef]
- Maity, S.; Saha, A. Therapeutic Potential of Exploiting Autophagy Cascade Against Coronavirus Infection. Front. Microbiol. 2021, 12, 675419. [Google Scholar] [CrossRef]
- Kang, C.K.; Seong, M.-W.; Choi, S.-J.; Kim, T.S.; Choe, P.G.; Song, S.H.; Kim, N.-J.; Park, W.B.; Oh, M.-D. In vitro activity of lopinavir/ritonavir and hydroxychloroquine against severe acute respiratory syndrome coronavirus 2 at concentrations achievable by usual doses. Korean J. Intern. Med. 2020, 35, 782–787. [Google Scholar] [CrossRef]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral Potential of ERK/MAPK and PI3K/AKT/mTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef]
- Shetty, R.M.; Namachivayam, A. Evidence for Chloroquine/Hydroxychloroquine in the Treatment of COVID-19. Indian J. Crit. Care Med. 2021, 25, 441–452. [Google Scholar] [CrossRef]
- Coombs, K.; Mann, E.; Edwards, J.; Brown, D.T. Effects of chloroquine and cytochalasin B on the infection of cells by Sindbis virus and vesicular stomatitis virus. J. Virol. 1981, 37, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Poschet, J.; Perkett, E.; Timmins, G.; Deretic, V. Azithromycin and ciprofloxacin Have A Chloroquine-Like Effect on Respiratory Epithelial Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cao, R.; Hu, H.; Li, Y.; Wang, X.; Xu, M.; Liu, J.; Zhang, H.; Yan, Y.; Zhao, L.; Li, W. Anti-SARS-CoV-2 Potential of Artemisinins in vitro. ACS Infect. Dis. 2020, 6, 2524–2531. [Google Scholar] [CrossRef]
- Sehailia, M.; Chemat, S. Antimalarial-agent Artemisinin and Derivatives Portray More Potent Binding to Lys353 and Lys31-binding Hotspots of SARS-CoV-2 Spike Protein than Hydroxychloroquine: Potential Repurposing of Artenimol for COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 6184–6194. [Google Scholar] [CrossRef]
- Uzun, T.; Toptas, O. Artesunate: Could be an Alternative Drug to Chloroquine in COVID-19 Treatment? Chin. Med. 2020, 15, 54. [Google Scholar] [CrossRef]
- Coleman, C.M.; Sisk, J.M.; Mingo, R.M.; Nelson, E.A.; White, J.M.; Frieman, M.B. Abelson Kinase Inhibitors Are Potent Inhibitors of Severe Acute Respiratory Syndrome Coronavirus and Middle East Respiratory Syndrome Coronavirus Fusion. J. Virol. 2016, 90, 8924–8933. [Google Scholar] [CrossRef]
- Yu, F.; Pan, T.; Huang, F.; Ying, R.; Liu, J.; Fan, H.; Zhang, J.; Liu, W.; Lin, Y.; Yuan, Y.; et al. Glycopeptide Antibiotic Teicoplanin Inhibits Cell Entry of SARS-CoV-2 by Suppressing the Proteolytic Activity of Cathepsin L. Front. Microbiol. 2021, 13, 1–17. [Google Scholar] [CrossRef]
- Baron, S.A.; Devaux, C.; Colson, P.; Raoult, D.; Rolain, J.-M. Teicoplanin: An alternative drug for the treatment of COVID-19? Int. J. Antimicrob. Agents 2020, 55, 105944. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.-F. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J. Infect. Public Health 2016, 9, 227–230. [Google Scholar] [CrossRef]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J.; et al. Personalized Therapeutic Cocktail of Wild Environmental Phages Rescues Mice from Acinetobacter baumannii Wound Infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef]
- Spriggs, M.K.; Collins, P.L. Human parainfluenza virus type 3: Messenger RNAs, polypeptide coding assignments, intergenic sequences, and genetic map. J. Virol. 1986, 59, 646–654. [Google Scholar] [CrossRef]
- Farahmand, M.; Malekshahi, S.S.; Jabbari, M.R.; Shayestehpour, M. The landscape of extrapulmonary manifestations of human parainfluenza viruses: A systematic narrative review. Microbiol. Immunol. 2020, 65, 1–9. [Google Scholar] [CrossRef]
- Gu, Y.E.; Park, J.Y.; Lee, M.K.; Lim, I.S. Characteristics of human parainfluenza virus type 4 infection in hospitalized children in Korea. Pediatr. Int. 2019, 62, 52–58. [Google Scholar] [CrossRef]
- Fathima, S.; Simmonds, K.; Invik, J.; Scott, A.N.; Drews, S. Use of laboratory and administrative data to understand the potential impact of human parainfluenza virus 4 on cases of bronchiolitis, croup, and pneumonia in Alberta, Canada. BMC Infect. Dis. 2016, 16, 402. [Google Scholar] [CrossRef]
- Abedi, G.R.; Prill, M.M.; Langley, G.E.; Wikswo, M.E.; Weinberg, G.A.; Curns, A.T.; Schneider, E. Estimates of Parainfluenza Virus-Associated Hospitalizations and Cost Among Children Aged Less Than 5 Years in the United States, 1998–2010. J. Pediatr. Infect. Dis. Soc. 2014, 5, 7–13. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome Supports Coxsackievirus B3 Replication in Host Cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Tang, Q.; Gao, P.; Arzberger, T.; Höllerhage, M.; Herms, J.; Höglinger, G.; Koeglsperger, T. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Estrabaud, E.; De Muynck, S.; Asselah, T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J. Hepatol. 2011, 55, 1150–1153. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Berryman, S.; Brooks, E.; Burman, A.; Hawes, P.; Roberts, R.; Netherton, C.; Monaghan, P.; Whelband, M.; Cottam, E.; Elazar, Z.; et al. Foot-and-Mouth Disease Virus Induces Autophagosomes during Cell Entry via a Class III Phosphatidylinositol 3-Kinase-Independent Pathway. J. Virol. 2012, 86, 12940–12953. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell. Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The Matrix Protein of Measles Virus Regulates Viral RNA Synthesis and Assembly by Interacting with the Nucleocapsid Protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, C.; Zhang, K.; Lu, N.; He, Y.; Liu, J.; Yang, Z.; Zhang, G. Identification of the Functional Domain of HPIV3 Matrix Protein Interacting with Nucleocapsid Protein. BioMed Res. Int. 2020, 2020, 2616172. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Sun, K.; Li, C.; Liao, S.; Yao, X.; Ouyang, Y.; Liu, Y.; Wang, Z.; Li, Z.; Yao, F. Ferritinophagy, a form of autophagic ferroptosis: New insights into cancer treatment. Front. Pharmacol. 2022, 13, 1043344. [Google Scholar] [CrossRef]
- Wang, J.; Wu, N.; Peng, M.; Oyang, L.; Jiang, X.; Peng, Q.; Zhou, Y.; He, Z.; Liao, Q. Ferritinophagy: Research advance and clinical significance in cancers. Cell Death Discov. 2023, 9, 463. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef]
- Nishio, M.; Tsurudome, M.; Ito, M.; Garcin, D.; Kolakofsky, D.; Ito, Y. Identification of Paramyxovirus V Protein Residues Essential for STAT Protein Degradation and Promotion of Virus Replication. J. Virol. 2005, 79, 8591–8601. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Yamaguchi, M.; Zhou, M.; Nishio, M.; Itoh, M.; Gotoh, B. Human Parainfluenza Virus Type 2 V Protein Inhibits TRAF6-Mediated Ubiquitination of IRF7 To Prevent TLR7- and TLR9-Dependent Interferon Induction. J. Virol. 2013, 87, 7966–7976. [Google Scholar] [CrossRef]
- Ohta, K.; Matsumoto, Y.; Nishio, M. Human parainfluenza virus type 2 V protein inhibits caspase-1. J. Gen. Virol. 2018, 99, 501–511. [Google Scholar] [CrossRef]
- Ohta, K.; Saka, N.; Nishio, M. Human Parainfluenza Virus Type 2 V Protein Modulates Iron Homeostasis. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghosh, S.; Mandal, A.; Ghosh, N.; Sil, P.C. ROS-associated immune response and metabolism: A mechanistic approach with implication of various diseases. Arch. Toxicol. 2020, 94, 2293–2317. [Google Scholar] [CrossRef]
- Lee, J.; Choi, E.H.; Lee, H.J. Comprehensive serotyping and epidemiology of human adenovirus isolated from the respiratory tract of Korean children over 17 consecutive years (1991–2007). J. Med Virol. 2010, 82, 624–631. [Google Scholar] [CrossRef]
- Lynch, J.P.; Fishbein, M.; Echavarria, M. Adenovirus. Semin. Respir. Crit. Care. Med. 2011, 32, 494–511. [Google Scholar] [CrossRef]
- Ison, M.G. Adenovirus Infections in Transplant Recipients. Clin. Infect. Dis. 2006, 43, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Barnadas, C.; Schmidt, D.J.; Fischer, T.K.; Fonager, J. Molecular epidemiology of human adenovirus infections in Denmark, 2011–2016. J. Clin. Virol. 2018, 104, 16–22. [Google Scholar] [CrossRef]
- Bil-Lula, I.; Ussowicz, M.; Rybka, B.; Wendycz-Domalewska, D.; Ryczan, R.; Gorczyńska, E.; Kałwak, K.; Woźniak, M. Hematuria due to Adenoviral Infection in Bone Marrow Transplant Recipients. Transplant Proc. 2010, 42, 3729–3734. [Google Scholar] [CrossRef] [PubMed]
- Kojaoghlanian, T.; Flomenberg, P.; Horwitz, M.S. The impact of adenovirus infection on the immunocompromised host. Rev. Med Virol. 2003, 13, 155–171. [Google Scholar] [CrossRef]
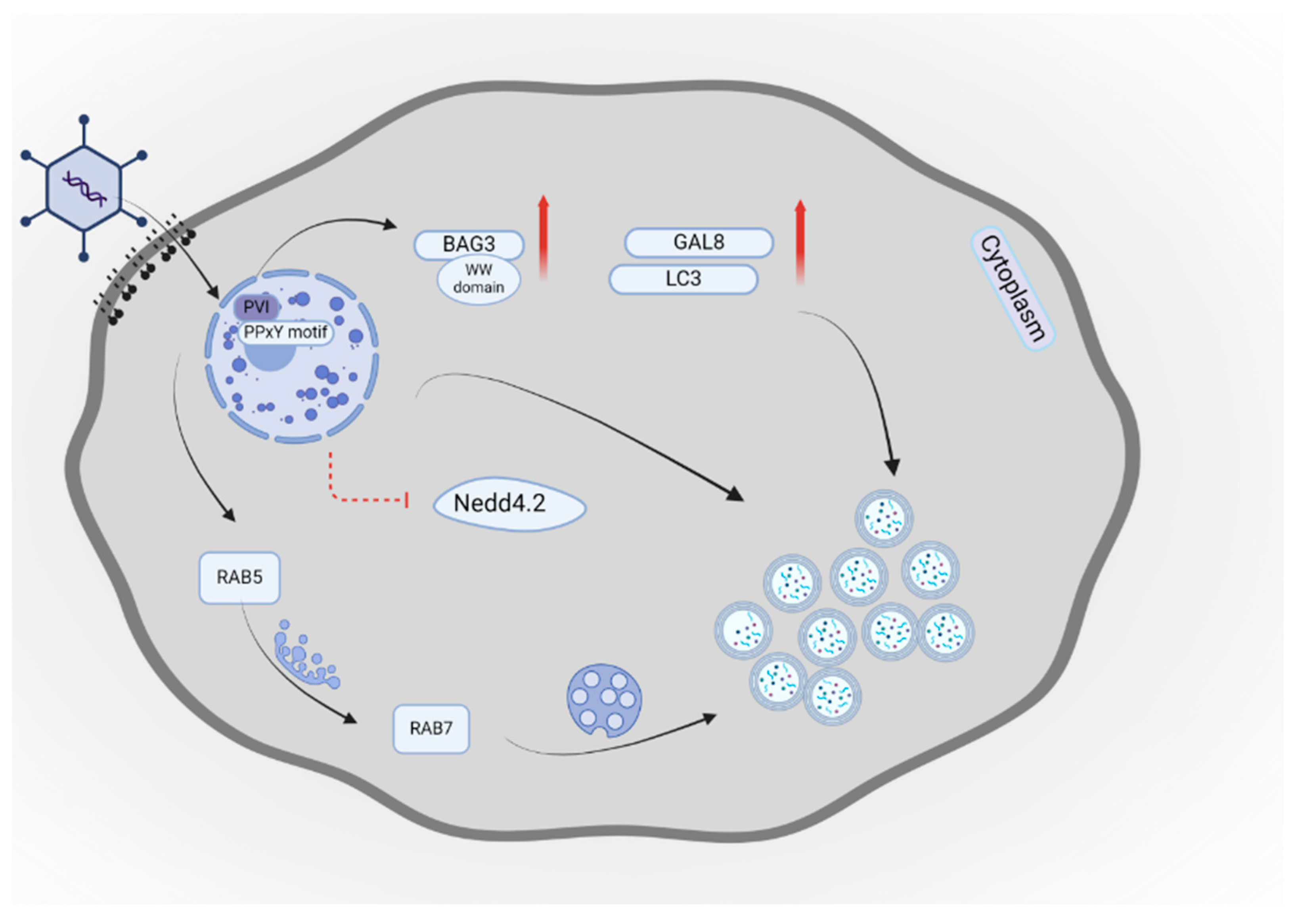
- Montespan, C.; Wiethoff, C.M.; Wodrich, H. A Small Viral PPxY Peptide Motif To Control Antiviral Autophagy. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A Capsid-Encoded PPxY-Motif Facilitates Adenovirus Entry. PLoS Pathog. 2010, 6, e1000808. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Li, G.; Marlin, M.C. Rab Family of GTPases. Methods Mol. Biol. 2015, 1298, 1–15. [Google Scholar]
- Girard, E.; Chmiest, D.; Fournier, N.; Johannes, L.; Paul, J.; Vedie, B.; Lamaze, C. Rab7 Is Functionally Required for Selective Cargo Sorting at the Early Endosome. Traffic 2014, 15, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Zeng, X.; Carlin, C.R. Host Cell Autophagy Modulates Early Stages of Adenovirus Infections in Airway Epithelial Cells. J. Virol. 2013, 87, 2307–2319. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, J.; Shen, L.; Chen, M.; Sun, K.; Li, J.; Li, M.; Ma, C.; Wei, L. Rab5c promotes RSV and ADV replication by autophagy in respiratory epithelial cells. Virus Res. 2024, 341, 199324. [Google Scholar] [CrossRef]
- Montespan, C.; Marvin, S.A.; Austin, S.; Burrage, A.M.; Roger, B.; Rayne, F.; Faure, M.; Campell, E.M.; Schneider, C.; Reimer, R.; et al. Multi-layered Control of Galectin-8 Mediated Autophagy During Adenovirus Cell Entry Through a Conserved PPxY Motif in the Viral Capsid. PLoS Pathog. 2017, 13, e1006217. [Google Scholar] [CrossRef]
- Zhang, L.; Duan, Y.; Wang, W.; Li, Q.; Tian, J.; Zhu, Y.; Wang, R.; Xie, Z. Autophagy induced by human adenovirus B7 structural protein VI inhibits viral replication. Virol. Sin. 2023, 38, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Stürner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Grand, A.R.J. The structure and functions of the adenovirus early region 1 proteins. Biochem. J. 1987, 241, 25–38. [Google Scholar] [CrossRef]
- Gheitasi, H.; Sabbaghian, M.; Fadaee, M.; Mohammadzadeh, N.; Shekarchi, A.A.; Poortahmasebi, V. The Relationship Between Autophagy and Respiratory Viruses. Arch. Microbiol. 2024, 206, 136. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.M.C.; Ribeiro, J.S.; da Silva, G.P.D.; da Costa, L.J.; Travassos, L.H.; Microbiology, I. Autophagy Modulators in Coronavirus Diseases: A Double Strike in Viral Burden and Inflammation. Front. Cell Infect. Microbiol. 2022, 12, 845368. [Google Scholar] [CrossRef]
- Horie, R.; Nakamura, O.; Yamagami, Y.; Mori, M.; Nishimura, H.; Fukuoka, N.; Yamamoto, T. Apoptosis and antitumor effects induced by the combination of an mTOR inhibitor and an autophagy inhibitor in human osteosarcoma MG63 cells. Int. J. Oncol. 2015, 48, 37–44. [Google Scholar] [CrossRef]
- Mullen, P.J.; Garcia, G., Jr.; Purkayastha, A.; Matulionis, N.; Schmid, E.W.; Momcilovic, M.; Sen, C.; Langerman, J.; Ramaiah, A.; Shackelford, D.B. SARS-CoV-2 Infection Rewires Host Cell Metabolism and is Potentially Susceptible to mTORC1 Inhibition. Nat. Commun. 2021, 12, 1876. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J.; et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Zinter, M. Azithromycin Treatment for Respiratory Syncytial Virus-Induced Respiratory Failure in Children, 2022. UCSF Clinical Trials. Available online: https://clinicaltrials.ucsf.edu/trial/NCT05026749#:~:text=The%20proposed%20study%20will%20be%20a%20randomized%2C%20double-blinded%2C,admission%20to%20the%20ICU%20at%2010%20pediatric%20hospitals (accessed on 10 December 2024).
- Huang, P.-J.; Chiu, C.-C.; Hsiao, M.-H.; Yow, J.; Tzang, B.-S.; Hsu, T.-C. Potential of antiviral drug oseltamivir for the treatment of liver cancer. Int. J. Oncol. 2021, 59, 109. [Google Scholar] [CrossRef]
- University of Oxford, Adaptive Assessment of Treatments for Influenza: A Phase 2 Multi-Centre Adaptive Randomised Platform Trial to Assess Antiviral Pharmacodynamics in Early Symptomatic Influenza Infection (AD ASTRA). 2023. Available online: https://www.medifind.com/articles/clinical-trial/389760524 (accessed on 10 December 2024).
- Gust, W. The Pharmacodynamic Functions of Low-Dose Rapamycin as a Model for Universal Influenza Protection. Undergraduate Thesis, University of Mississippi, University, MS, USA, 2017. Available online: https://egrove.olemiss.edu/hon_thesis/949/ (accessed on 10 December 2024).
- Huang, C.-T.; Hung, C.-Y.; Chen, T.-C.; Lin, C.-Y.; Lin, Y.-C.; Chang, C.-S.; He, Y.-C.; Huang, Y.-L.; Dutta, A. Rapamycin adjuvant and exacerbation of severe influenza in an experimental mouse model. Sci. Rep. 2017, 7, 4136. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.C. Adjunctive Sirolimus and Oseltamivir Versus Oseltamivir Alone for Treatment of Influenza, 2023. Good Clinical Practice Network. Available online: https://ichgcp.net/clinical-trials-registry/NCT03901001 (accessed on 10 December 2024).
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjós, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63. [Google Scholar] [CrossRef]
- University of Chicago, SECOVID: A Multi-Center, Randomized, Dose-Ranging Parallel-Group Trial Assessing the Efficacy of Sirolimus in Hospitalized Patients with COVID-19 Pneumonia for the Prevention of Post-COVID Fibrosis. 2021. FDAAA TrailsTracker. Available online: https://fdaaa.trialstracker.net/trial/NCT04948203/ (accessed on 10 December 2024).
- Mondaca-Ruff, D.; Riquelme, J.A.; Quiroga, C.; Norambuena-Soto, I.; Sanhueza-Olivares, F.; Villar-Fincheira, P.; Hernández-Díaz, T.; Cancino-Arenas, N.; San Martin, A.; García, L.; et al. Angiotensin II-regulated Autophagy is Required for Vascular Smooth Muscle Cell Hypertrophy. Front. Pharmacol. 2019, 9, 1553. [Google Scholar] [CrossRef]
- Puigdellívol-Sánchez, A. Evaluation of Influenza Vaccination and Treatment with ACEI and ARB in the Evolution of SARS-CoV2 Infection. 2020. Available online: https://www.medifind.com/articles/clinical-trial/221191378 (accessed on 10 December 2024).
- Brimson, J.M.; Prasanth, M.I.; Malar, D.S.; Brimson, S.; Thitilertdecha, P.; Tencomnao, T. Drugs that Offer the Potential to Reduce Hospitalization and Mortality from SARS-CoV-2 Infection: The Possible Role of the Sigma-1 Receptor and Autophagy. Expert Opin. Ther. Targets 2021, 25, 435–449. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Family | Mechanisms |
|---|---|---|
| Influenza A virus (IAV) | Orthomyxoviridae | M2 ion channel blocks autophagosome–lysosome fusion [19,20] |
| SARS-CoV-2 | Coronaviridae | ORF3a blocks autophagosome–lysosome fusion; NSP6 limits autophagosome expansion [21,22] |
| Respiratory Syncytial Virus (RSV) | Pneumoviridae | NS2 protein activates autophagy through BECN1 [23,24] |
| Parainfluenza Virus (PIV) | Paramyxoviridae | Phosphoprotein P induces incomplete autophagy, blocking autophagosome–lysosome fusion [25,26] |
| Adenovirus | Adenoviridae | E1B-19K protein interacts with BECN1 to regulate autophagy [27,28] |
| Agent | Impact on influenza infection | Impact on autophagy | References |
|---|---|---|---|
| Gallic acid | Decreases viral load | Reduces accumulation of autophagosomes | [86] |
| Vitamin D3 | Induces cytoprotective effects | Enhances fusion of autophagosome and lysosome, thus decreasing viral replication | [87] |
| Baicalin | Improves viability of infected macrophages | Reduces expression of autophagy marker | [88] |
| Tanreqing | Inhibits influenza replication | Enhances fusion of autophagosome and lysosome | [65] |
| Huanglian-Ganjiang combination | Suppresses inflammatory responses | Enhances fusion of autophagosome and lysosome | [89] |
| Category | Medications | Mechanism | References |
|---|---|---|---|
| Lysosomotropic Agents | Chloroquine, Hydroxychloroquine | Increases the pH within lysosomes/blocks entry mechanisms of virus/does not inhibit infection of human lung cells with SARS-CoV-2; also blocks some viruses’ biosynthetic processes after entry | [161,166,167] |
| Azithromycin | Synergistic effect of hydroxychloroquine and azithromycin on the reduction of viral load of SARS-CoV-2 | [168] | |
| Artemisinin | Targets the Lys353 and Lys31 binding hotspots on the viral spike protein/NF-κB inhibition/blocks SARS-CoV-2 infection | [169,170,171] | |
| Imatinib | Inhibits fusion of the virions at the endosomal membrane | [172] | |
| Protease Inhibitors | Lopinavir/Ritonavir | Inhibits viral protease/reduction in viral load | [163,164] |
| Teicoplanin | Suppresses the proteolytic activity of cathepsin L on Spike/prevents the entry of SARS-CoV-2 into the cytoplasm | [173,174] | |
| PI3K/mTOR Regulators | Rapamycin | Inhibits mTORC1/inhibits protein synthesis/reduces viral replication/reduces MERS-CoV and SARS-CoV-2 infection by activating autophagy | [115] |
| Everolimus | Induces autophagy by blocking mTORC1/inhibits MERS-CoV infection | [165] | |
| Nitazoxanide | Stimulates autophagy by blocking mTORC1/inhibits replication of MERS-CoV and SARS-CoV-2 | [175,176] | |
| Wortmannin | Suppresses autophagy by inhibition of PI3K/inhibits MERS-CoV infection | [165] |
| Trial Identifier | Activity | Intervention | Phase | Primary Outcome | Link |
|---|---|---|---|---|---|
| NCT05060705 | COVID-19 | Efesovir in comparison with the drug Remdesivir | Phase 2 | Reduction of viral load in COVID-19 patients | https://clinicaltrials.gov/study/NCT05060705 (Accession date: 10 December 2024) |
| NCT05218356 | COVID-19 | Codivir | Phase 2 | Efficacy in reducing the severity of COVID-19 | https://clinicaltrials.gov/study/NCT05218356 (Accession date: 10 December 2024) |
| NCT06128967 | COVID-19 | Metformin/Fluvoxamine | Phase 3 | Evaluation of treatment efficacy in long COVID patients | https://clinicaltrials.gov/study/NCT06128967 (Accession date: 10 December 2024) |
| NCT06147050 | COVID-19 | Metformin | Phase 3 | Assessment of Chronic Fatigue Syndrome in long COVID patients | https://clinicaltrials.gov/study/NCT06147050 (Accession date: 10 December 2024) |
| NCT04345406 | COVID-19 | ACE inhibitors | Phase 3 | Clinical efficacy in COVID-19 treatment | https://clinicaltrials.gov/study/NCT04345406 (Accession date: 10 December 2024) |
| NCT04948203 | COVID-19 | Sirolimus | Phase2 Phase 3 | Prevention of post-COVID-19 fibrosis in hospitalized patients | https://clinicaltrials.gov/study/NCT04948203 (Accession date: 10 December 2024) |
| NCT06024096 | Influenza | Atorvastatin | Phase 4 | Effect of statins on influenza vaccine response | https://clinicaltrials.gov/study/NCT06024096 (Accession date: 10 December 2024) |
| NCT05026749 | RSV | Azithromycin | Phase 3 | Efficacy in RSV-induced respiratory failure in children | https://clinicaltrials.gov/study/NCT05026749 (Accession date: 10 December 2024) |
| NCT03901001 | Influenza | Sirolimus + Oseltamivir vs. Oseltamivir Alone | Phase 3 | Comparison of treatment outcomes for influenza | https://clinicaltrials.gov/study/NCT03901001 (Accession date: 10 December 2024) |
| NCT05648448 | Influenza | Influenza antivirals | Phase 2 | Assessing antiviral efficacy in early symptomatic influenza | https://clinicaltrials.gov/study/NCT05648448 (Accession date: 10 December 2024) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aligolighasemabadi, F.; Bakinowska, E.; Kiełbowski, K.; Sadeghdoust, M.; Coombs, K.M.; Mehrbod, P.; Ghavami, S. Autophagy and Respiratory Viruses: Mechanisms, Viral Exploitation, and Therapeutic Insights. Cells 2025, 14, 418. https://doi.org/10.3390/cells14060418
Aligolighasemabadi F, Bakinowska E, Kiełbowski K, Sadeghdoust M, Coombs KM, Mehrbod P, Ghavami S. Autophagy and Respiratory Viruses: Mechanisms, Viral Exploitation, and Therapeutic Insights. Cells. 2025; 14(6):418. https://doi.org/10.3390/cells14060418
Chicago/Turabian StyleAligolighasemabadi, Farnaz, Estera Bakinowska, Kajetan Kiełbowski, Mohammadamin Sadeghdoust, Kevin M. Coombs, Parvaneh Mehrbod, and Saeid Ghavami. 2025. "Autophagy and Respiratory Viruses: Mechanisms, Viral Exploitation, and Therapeutic Insights" Cells 14, no. 6: 418. https://doi.org/10.3390/cells14060418
APA StyleAligolighasemabadi, F., Bakinowska, E., Kiełbowski, K., Sadeghdoust, M., Coombs, K. M., Mehrbod, P., & Ghavami, S. (2025). Autophagy and Respiratory Viruses: Mechanisms, Viral Exploitation, and Therapeutic Insights. Cells, 14(6), 418. https://doi.org/10.3390/cells14060418