1. Introduction
Breast cancer is a major health challenge affecting millions of women worldwide [
1]. While advancements in breast cancer prognosis and treatment have made significant progress, the complexity of breast cancer outcomes is still influenced by factors such as tumor heterogeneity. Tumor cell heterogeneity is thought to be maintained and promoted by CSCs, a sub-population of cancer cells that are capable of self-renewing and differentiating [
2,
3,
4]. CSCs are a subpopulation of cells within a tumor that possess characteristics similar to normal stem cells. They are capable of self-renewal, differentiation, and sustaining tumor growth. These cells play a critical role in cancer progression including metastasis, resistance to therapy, and recurrence [
3,
5,
6]. Understanding the properties and functional activities of CSCs, as well as the regulatory mechanism of CSCs hold promise for developing novel therapeutic strategies and improving efficacy of breast cancer treatment [
7].
Cancer cells are highly dependent on glycolysis to meet the demand for rapid proliferation and tumor growth [
8]. However, beside the major influx of glucose being consumed in glycolysis, a small percentage of glucose entering cancer cells is used in the hexosamine biosynthesis pathway (HBP), which utilizes glucose and other products of major metabolic pathways to form UDP-N-acetylglucosamine (UDP-GlcNAc) [
9]. UDP-GlcNAc serves as the substrate for N-linked and O-linked glycosylation of membrane and secreted proteins [
10], and for modification of intracellular proteins in a process called O-GlcNAcylation [
11]. This modification modulates protein stability, protein-protein interaction, protein phosphorylation and protein localization [
12]. Both O-GlcNAcylation and its enzyme O-GlcNAc transferase (OGT) are highly expressed in many cancers, where it promotes tumor growth [
13,
14] and metastasis [
15,
16]. Importantly, recent studies have implicated the role of elevated OGT and O-GlcNAc in regulating CSCs phenotypes and tumor initiation [
17]. However, the mechanism by which OGT and O-GlcNAc regulate CSCs in cancer is not clear.
The Nucleosome Remodeling and Deacetylase complex (NuRD complex) is a multi-protein complex that participates in regulation of gene expression by restructuring chromatin architecture and by deacetylation of histone. The NuRD complex plays a pivotal role in maintaining cellular homeostasis, regulating gene expression during development, differentiation, and DNA repair [
18]. The complex is also critical for stem cell function and progenitor cells activities [
19,
20]. A number of components of the NuRD complex have been linked to cancer, including GATAD2B, a key scaffold-protein in the complex [
21,
22]. However, the function of GATAD2B in breast cancer has not yet been explored. Here, we show for the first time that GATAD2B is critical in maintaining and promoting CSCs phenotypes while O-GlcNAcylation of GATAD2B enhances its stability and protects from ITCH-mediated proteasomal degradation and contributes to cancer stem cell functions in breast cancer cells.
2. Materials and Methods
2.1. Cell Lines
Human TNBC cells MDA-MB-231 were purchased from the ATCC (American Type Culture Collection, Manassas, VA, USA), and were cultured in humidified condition with 5% CO2 in complete medium Dulbecco’s Modification Eagle’s Medium with 4.5 g/L glucose (DMEM, Genesee Scientific, El Cajon, CA, USA), supplemented with 10% fetal bovine serum (FBS, GeminiBio, West Sacramento, CA, USA), 1% L-Glutamine (Gibco/ThermoFisher Scientific, Waltham, MA, USA), 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). TNBC cells SUM159 and patient-derived xenograft TNBC cells HCI-10 were received as a kind gift from Dr. Seagroves (University of Tennessee). SUM159 cells were maintained in DMEM/F12 medium with 10% FBS (Gemini), 1% pencicillin/streptomycin (Invitrogen), 1% L-Glutamine (Gibco) and 0.05% Hydrocortisone. HCI-10 cells were cultured in DMEM/F12 medium with 2% of low endotoxin FBS (Gibco), 1× penicillin/streptomycin (Gibco), 1× Insulin-transferrin-selenium, 5 ng/mL hEGF, 0.5 ng/mL chorela toxin, 0.3 μg/mL hydrocortisone, 5 nM 3,3′,5-triiodo-L-thyronine, 5 μM isoproterenol hydrochloride, 50 nM ethanolamine, 50 nM O-phosphorylethanolamine and 10 mM HEPES. HEK-293T cell was purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and was cultured in humidified condition at 37 °C with 5% CO2 using Dulbecco’s Modification Eagle’s Medium with 4.5 g/L glucose (DMEM, Genesee) with 10% FBS (Gemini), 1% L-Glutamine (Gibco/Life-Technologies), and 1% penicillin/streptomycin (Invitrogen). Quarterly testing was conducted to ensure that all the cells remained free from mycoplasma contamination (abmGood PCR Mycoplasma Detection Kit, Richmond, BC, Canada).
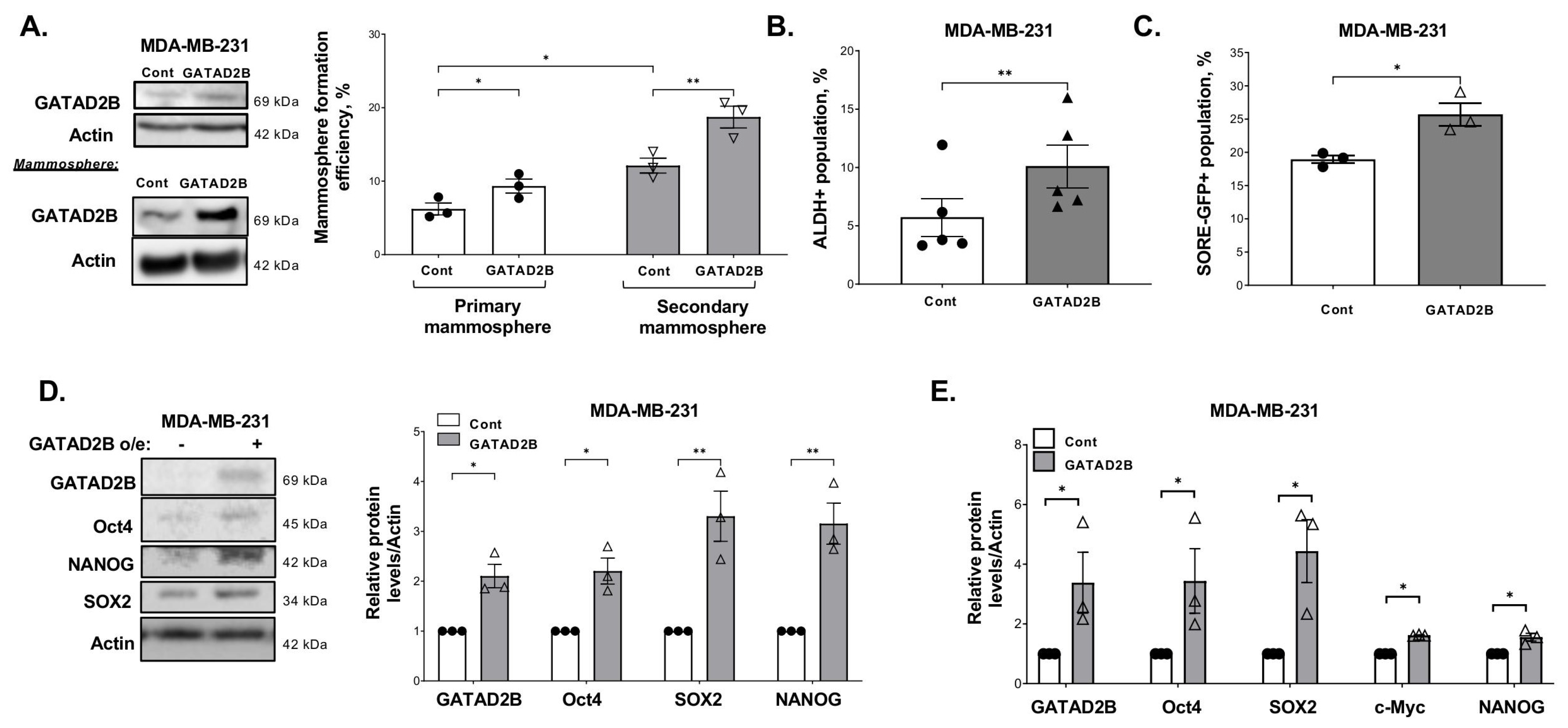
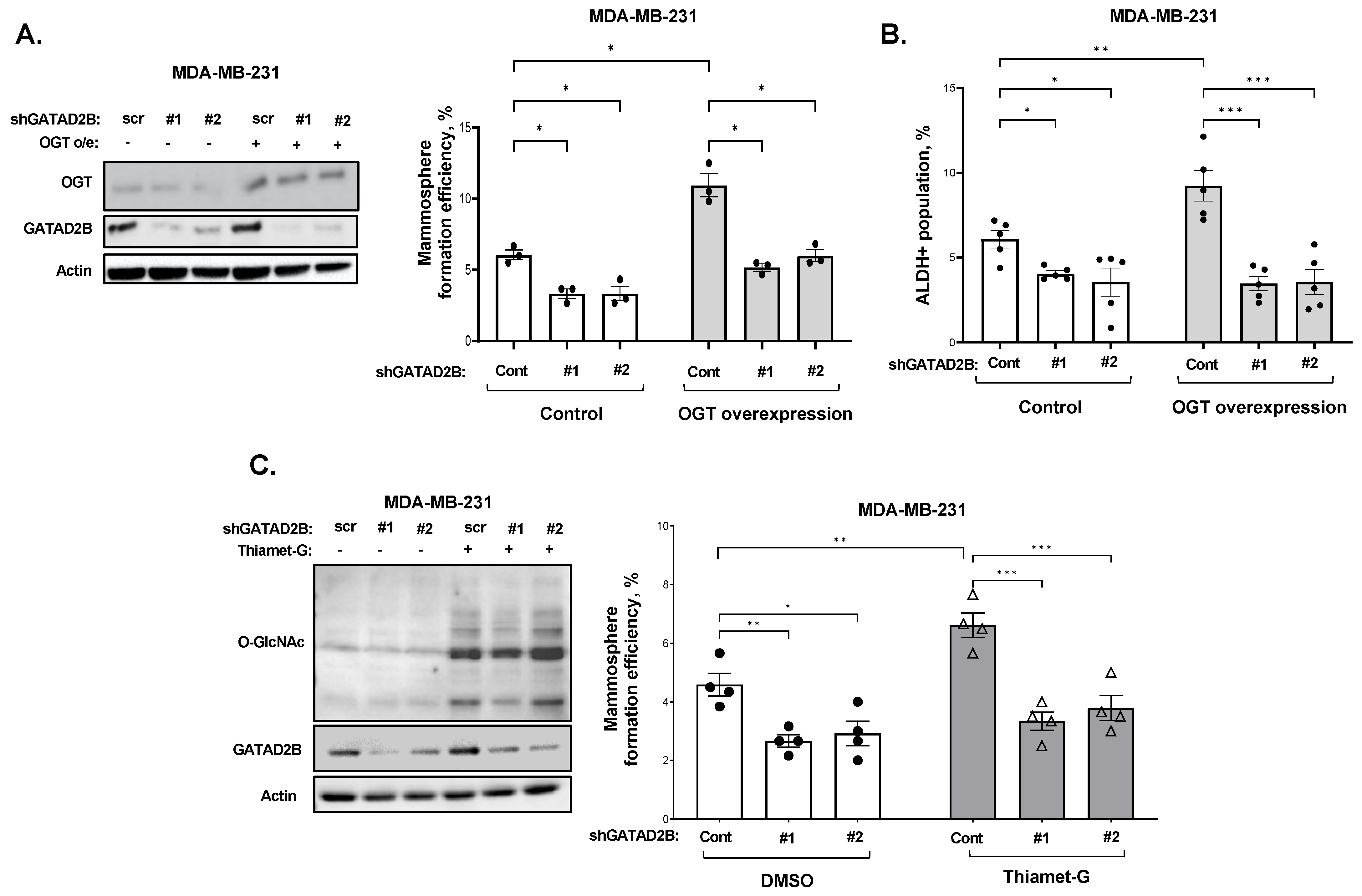
2.2. Generating Cells with GATAD2B or OGT Overexpression
To generate TNBC cells stably overexpressing GATAD2B or OGT, lentiviral transduction was performed as previously described [
17] using the respective lentiviral vectors obtained from GeneCopoeia, Rockville, MD, USA (EX-E0581-Lv118-GATAD2B and EX-Z3428-Lv101-OGT). Construct containing GATAD2B S584/586/588/590A mutant was generated and obtained from GeneCopoeia based on the template of EX-E0581-Lv118-GATAD2B.
2.3. Transient Transfection of Mammalian Expression Constructs
Transient transfection was conducted in HEK293T cells, which were cultured in the condition described above. Transfection with PEI was performed as described before using 15 µg of DNA construct. pRK5-HA-Ub was purchased from Addgene, Watertown, MA, USA, from Dr. Dawson (John Hopkins University School of Medicine). Wild-type HA-tagged GATAD2B and mutant GATAD2B (S584/586/588/590A) was obtained from GeneCopoeia (EX-E0581-Lv118-GATAD2B). Protein expression was allowed to continue for 3 days after transfection, prior to further analysis.
2.4. RNA Interference
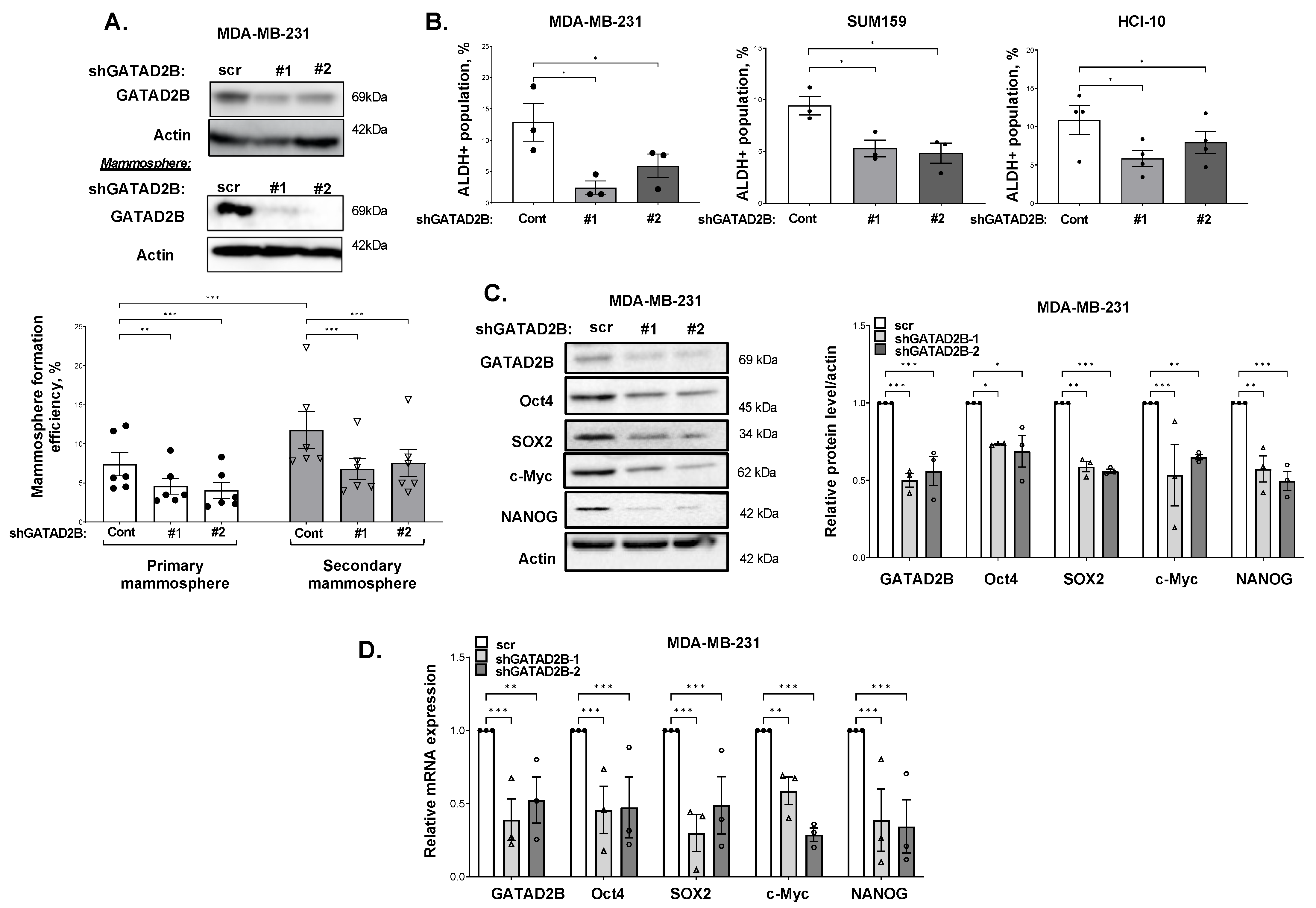
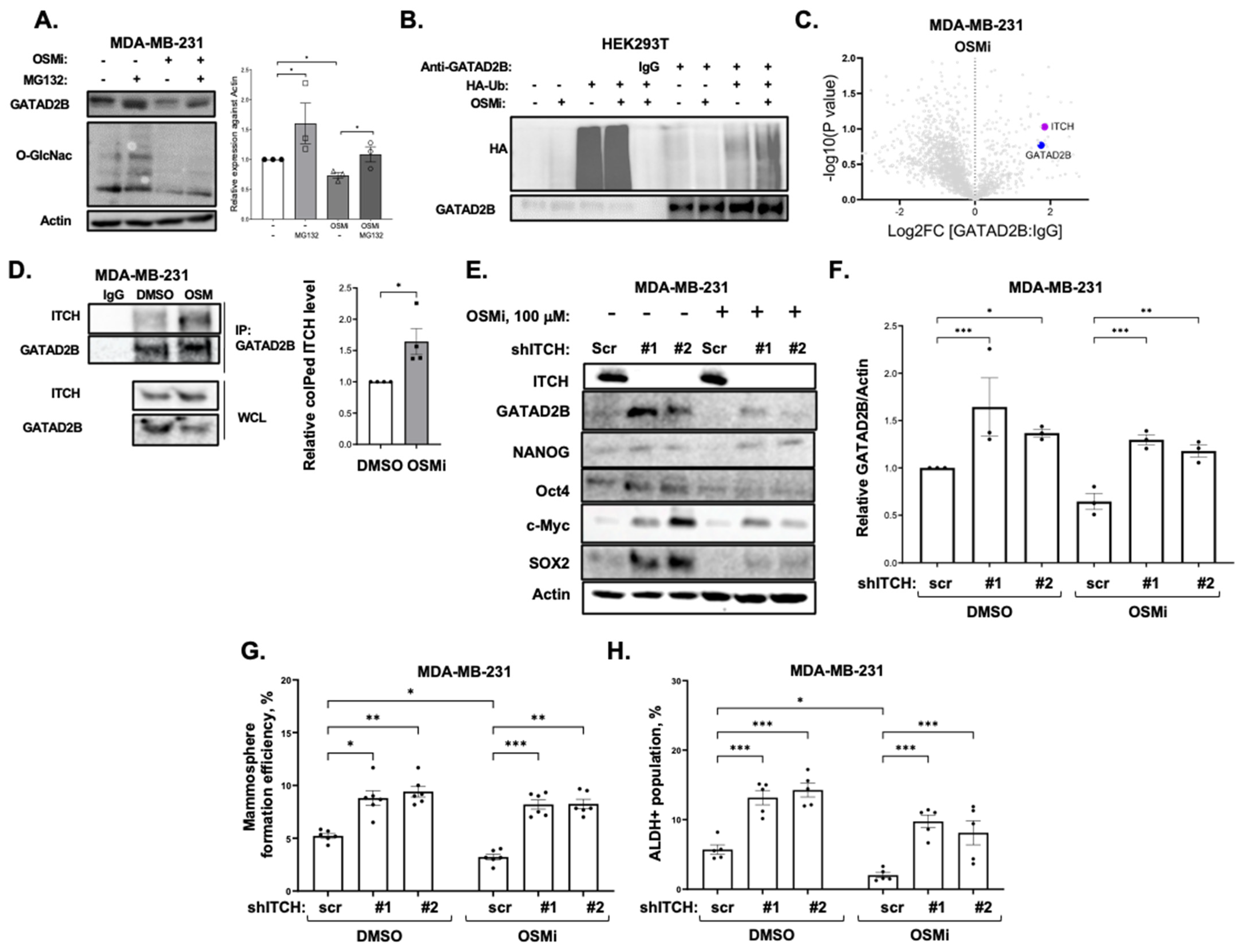
GATAD2B knockdown was performed using lentiviral vectors carrying shRNA constructs obtained from Sigma-Aldrich, St. Louis, MO, USA, (TRCN0000015315, TRCN0000015317). The sequences of used shRNA targeting GATAD2B are: 5′-CGCTCCATGCTTTCAAACTTT-3′ (shGATAD2B #1) and 5′-CAGGAAATTGAACAGCGATTA-3′ (shGATAD2B #2). ITCH knockdown was conducted using lentiviral shRNA constructs purchased from Sigma-Aldrich (TRCN0000002087, TRCN0000010680). The sequences of used shITCH constructs are: 5′-CCAGAAGTCAAGGTCAATTAA-3′ (shITCH #1) and 5′-CGAAGACGTTTGTGGGTGATT-3′ (shITCH #2). Control shRNA (plasmid 1864) was purchased from Addgene. OGT shRNA was obtained from Sigma-Aldrich. The sequence of used shRNA targeting OGT is: 5′-GCTGAGCAGTATTCCGAGAAACTCGAGTTTCTCGGAATACTGCTCAGC-3′ (shOGT). The sequence of used control shRNA is: 5′-CCTAAGGTTAAGTCGCC-CTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′. pLKO1-puro vectors carrying shRNA targeting GATAD2B, ITCH, OGT, or control shRNA were packaged into VSVG-pseudo-lentiviral particles using HEK-293T cells. Lentiviral transduction was conducted as described before to generate GATAD2B, ITCH or OGT knockdown.
CRISPR targeting GATAD2B was conducted using gRNA obtained from Sigma-Aldrich (CRISPRD HSPD0000102326) with the following sequence 5′-TCGCTTGAATCTGTTGAAG-3′. The control gRNA was also obtained from Sigma-Aldrich (CRISPRD NegativeControl1) with the following sequence 5′-CGCGATAGCGCGAATATATT-3′. All-in-one lentiviral vector LV01-U6-gRNA:ef1a-puro-2A-Cas9-2A-tGFP carrying gRNA targeting GATAD2B or control gRNA was packaged into VSVG-pseudo-lentiviral vector using HEK293T cells. Lentiviral transduction was conducted as described before.
2.5. Mammosphere Formation Assay
Breast cancer cells were collected and a fixed amount of cells (between 200–800 cells depending on mammosphere formation effeciency of specific cell line) was cultured in ultralow-attachment 96-well plate, pre-coated with 1.2% polyHEMA (Corning) as previously described [
17]. Seeded cells were allowed to form in DMEM/F12 (Gibco) medium, supplemented with 1 mg/mL Pen/Strep (Gibco), 1× B27 (Invitrogen, 17504-044), 20 ng/mL EGF (Sigma-Aldrich) and 20 ng/mL bFGF (Gibco, PHG0024). Mammospheres were allowed to form in humidified condition with 5% CO
2, at 37 °C for 5–7 days. The number of mammosphere greater than 50 µm was counted and mammosphere formation efficiency was determined using the following formula: (number of mammospheres/number of cells plated) * 100%.
For secondary mammosphere, breast cancer cells were allowed to grow in 6-well plate, coated with 1.2% polyHEMA in the same condition as described above. After 5–7 days, mammospheres were collected to prepare single-cell suspension. A fixed number of cells (200–800 cells) was then cultured in the mammosphere forming assay as described above. the same condition as described above. The efficiency of mammosphere formation was determined following the same formula as described above.
2.6. Flow Cytometry
ALDEFLOUR assay was performed using ALDEFLOUR assay kit purchased from STEMCELL Technologies, Vancouver, British Columbia, Canada, as per manufacturer instruction. In brief, breast cancer cells were harvested, washed in PBS twice, and resuspneded in ALDEFLOUR assay buffer at concentration of 2 × 105 cells/mL. 1 mL of cell suspension was then rapidly mixed with 5 μL of activated provided substrate BAAA-DA and 500 μL of the mixture was immediately taken out and mixed with 5 μL of ALDGH-specific inhibitor-DEAB. Samples with DEAB and without DEAB were then incubated at 37 °C for 30 min, followed by centrifugation to remove exceeded BAAA-DA.
The cell pellet was resuspended in 500 μL of ALDEFLOUR assay buffer. Fluorescence signal generated from ALDH activity in the presence of BAAA-DA was detected by flow cytometry using the system Guava-easyCyte (MilliporeSigma, Burlington, MA, USA). The basal fluorescence signal was determined using sample with ALDH inhibitor DEAB. The percentage of ALDH-positive cells represents the population of cancer cells with high ALDH activity.
SOX2/Oct4-Response element GFP (SORE-GFP) reporter was received as a kind gift from Dr. Wakefield (NCI) [
23]. Lentiviral vectors containing SORE-GFP construct were generated in HEK293T cells, and lentiviral transduction was conducted as described before. A construct containing SORE-GFP with minimal-CMV promoter also received from Dr. Wakefield (NCI) was used as a control. For SORE-GFP assay, breast cancer cells transduced with control or SORE-GFP construct were collected, washed in PBS, and counted. 2 × 10
5 cells from each sample was pelleted and resuspended in 500 μL PBS. The fluorescence emission from GFP was assessed by flow cytometry using the system Guava-easyCyte (Millipore). Basal fluorescence signal was determined using the control sample with minimal CMV promoter.
2.7. Western Blotting
Breast cancer cells were harvested, centrifuged, and thoroughly washed with ice-cold PBS. Cells were then lysed in RIPA buffer containing 150 mM NaCl, 1% NP40, 0.5% Deoxycholate, 50 mM Tris-HCl (pH 8), 0.1% SDS, 10% glycerol, 5 mM EDTA, 20 mM NaF, and 1 mM Na3VO4, supplemented with protease inhibitors. After lysis, all lysates were centrifuged at 15,000 rpm for 20 min at 4 °C and supernatants were collected for immunoblot analysis. Protein concentration was determined using Bradford assay. 50–100 ug of total proteins was separated by SDS-PAGE and transferred to PVDF membrane with 0.45 µm pore size. The following primary antibodies were used to detected proteins of interest: Anti-OGT (Cell-Signaling, Danvers, MA, USA, cat #20438), Anti-O-GlcNAc (Sigma, cat #MABS1254), Anti-c-MYC (Novus Biologicals, Centennial, CO, USA, cat #NB600-335), Anti-Actin (Santa Cruz, Dallas, TX, USA, cat #sc-47778), Anti-NANOG (Cell-Signaling, cat #3580), Anti-SOX2 (Cell-Signaling, cat #3579), Anti-OCT4 (Cell-Signaling, cat #2750), Anti-GATAD2B (Cell-Signaling, cat #73098), anti-GATAD2A (Cell-Signaling, cat #17705), anti-MTA2 (Sigma, cat #HPA006214), anti-CHD4 (Cell-Signaling, cat #12011), anti-HDAC2 (Cell-Signaling, cat #57156), anti-RBBP4 (Cell-Signaling, cat #9067), anti-ITCH (Cell-Signaling, cat #12117).
2.8. Mass Spectrometry Analysis
Effect of Thiamet G on protein expression. MDA-MB-231 cells were grown in the presence or absence of Thiamet G (Sigma-Aldrich) at a final concentration of 1 µM for 24 h. Cells were washed three times with cold PBS prior to harvesting and lysed with freshly prepared lysis buffer (9 M urea, 50 mM Tris (pH 8), and 100 units/mL Pierce Universal Nuclease (Thermo Fisher Scientific, Waltham, MA, USA)). The lysates were sonicated (Fisherbrand Model 120 Sonic Dismembrator, Fisher Scientific) by applying 20 s on/off pulses for 3 cycles and centrifuged at 16,000× g for 15 min to pellet cell debris. Protein concentration was determined by bicinchoninic acid protein assay (Pierce BCA Protein Assay Kit, Thermo Scientific). Proteins were reduced with 1 mM 1,4-Dithiothreitol (DTT) (Thermo Scientific) for 30 min at 25 °C followed by alkylation with 5.5 mM iodoacetamide (IAA) (Thermo Scientific) for 15 min at 25 °C in the dark. The urea concentration was diluted to ~1.6 M with 50 mM ammonium bicarbonate (pH 8.0) prior to sequential digestion with Lys-C (1:50 enzyme: protein ratio, Wako Tokyo, Japan) for 3 h at 25 °C and trypsin (1:50 enzyme: protein ratio, Sigma) at 37 °C for 18 h. Acidified samples were desalted using C18 StageTips (Thermo Scientific SP301) and dried by vacuum centrifugation. Peptides were separated and analyzed on an EASY nLC 1200 System (Thermo Scientific) in-line with the Orbitrap Fusion Lumos Mass Spectrometer (Thermo Scientific) with instrument control software v. 4.2.28.14. Two µg of tryptic peptides were pressure loaded onto C18 reversed phase column (Acclaim PepMap RSLC, 75 µm × 50 cm (C18, 2 µm, 100 Å) ThermoFisher cat. #164536) using a gradient of 5% to 35% B in 180 min (Solvent A: 5% acetonitrile/0.1% formic acid; Solvent B: 80% acetonitrile/0.1% formic acid) at a flow rate of 300 nL/min. Mass spectra were acquired in data-dependent mode with a high resolution (60,000) FTMS survey scan, mass range of m/z 375–1575, followed by tandem mass spectra (MS/MS) of the most intense precursors with a cycle time of 3 s. The automatic gain control target value was 4.0 × 105 for the survey MS scan. HCD fragmentation was performed with a precursor isolation window of 1.6 m/z, a maximum injection time of 50 ms, and collision energy of 35%. Monoisotopic-precursor selection was set to “peptide”. Precursors within 10 ppm mass tolerance were dynamically excluded from resequencing for 15 s. Precursor ions with charge states 2–5 were included. Data were searched against a forward and reversed, decoy human protein database downloaded from Uniprot on 4 February 2020 as well as a database of common cell culture contaminants using the MaxQuant (v 1.0.17.0) (Max Planck Institute). The search parameters allowed a maximum of two missed cleavages and a minimum peptide length of seven amino acids. Cysteine carbamidomethylation was set as a fixed modification while acetylation at protein N-termini and oxidation of methionines were set as variable modifications. Matching between runs was enabled. For identification an FDR < 0.01 was required at the PSM, peptide, and protein levels. Data were processed using Perseus v1.6.12.0 (Max Planck Institute). Proteins were filtered to remove matches to the reversed database, common contaminants, and proteins identified by a single modified peptide. Normalized protein intensities were log2 transformed. The proteins were filtered to retain those quantified in 3 biological replicates of control or TMG treated cells. Missing values were imputed from a normal distribution with a width of 0.3 and down shift of 1.8 from the total matrix. Log2 protein intensities from each condition were compared using a Student’s t-test. The log2 fold changes, p values, and q values are provided.
Identification of O-GlcNAc modified proteins. Protein extracts from thiamet G treated MDA-MB-231 cells were prepared as described above. Following trypsin digestion, N-linked glycans were removed with peptide-N-glycosidase F (PNGase F) PRIME (N-Zyme Scientifics, Doylestown, PA, USA), at 37 °C and 300 rpm for 18 h (1:100 enzyme:protein). The reaction was inactivated with formic acid (FA) to a final concentration of 1% followed by centrifugation at 16,000×
g for 10 min to remove debris. Peptides were desalted with 500 mg sorbent Sep-Pak tC18 6 cc cartridges (Waters WAT036790) according to the manufacturer’s instructions and dried under vacuum. For high pH RP fractionation, peptides were resuspended in 2 mL of 50 mM ammonium bicarbonate and separated at 3 mL/min on a Zorbax 300 Å Extend-C18 column (9.4 × 250 mm, 5 µm, 300 Å, Agilent Technologies, Santa Clara, CA, USA) using a gradient of 1–25% B in 50 min, 25–60% in 4 min, 60–70% in 2 min, and 70% B for 9 min (Solvent A: 50 mM ammonium bicarbonate; Solvent B: 10% 50 mM ammonium bicarbonate with 90% acetonitrile (
v/
v)). One hundred and twenty 1.5 mL fractions were collected, brought to neutral pH with 10% FA, concatenated into 10 fractions, and dried. Peptides were desalted with Sep-Pak tC18 3 cc cartridges (Waters), dried by vacuum centrifugation, and stored at −80 °C. O-GlcNAc modified peptides were immunoaffinity enriched using PTMScan O-GlcNAc [GlcNAc-S/T] Motif antibody kit (Cell Signaling, cat #95220) [
24]. Anti-O-GlcNAc antibody beads were washed and resuspended in 160 μL of cold immunoaffinity purification (IAP) buffer. Peptides (~1 mg/fraction) reconstituted at 1 mg/mL in IAP buffer were incubated with 40 uL beads for 2 h at 4 °C with end-to-end rotation. Samples were centrifuged and supernatants deposited into clean tubes for a sequential enrichment using fresh antibody beads. Beads from the two sequential immunoaffinity steps were washed and peptides eluted with 0.15% trifluoroacetic acid were combined. Peptides from each fraction were desalted twice using C18 StageTips (Thermo Scientific) as described with the PTMScan O-GlcNAc [GlcNAc-S/T] Motif Kit. Samples were dried by vacuum centrifugation and stored at −80 °C. LC-MS/MS was performed as above with the following changes in instrument parameters. MS2 spectra of ions with charge state 2–6 were acquired in data dependent mode with alternating HCD and ETD and a cycle time of 3 s. HCD spectra were collected at 15,000 MS2 resolution, AGC target of 1 × 10
5 (200% normalized AGC target), maximum ion injection time of 105 ms, and 40% collision energy. Electron transfer dissociation (ETD) spectra were acquired at 15,000 MS2 resolution, AGC target of 4 × 10
5 (800% normalized AGC target), and maximum ion injection time of 120 ms using charge-dependent reaction times. The raw files were searched using MaxQuant version 2.4.2.0 (Max Plank Institute) against a human protein database downloaded 02022024 from UniProt as well as a database of common cell culture contaminants. Variable modifications specified were: HexNAc modification or phosphorylation of Ser, Thr, Tyr [
25], oxidation of Met, and protein N-terminal acetylation. Within Andromeda, a customized modification was defined accounting for the neutral loss of GlcNAc and the diagnostic HexNAc oxonium ions (OGlcNAc_NL_Std). Identified peptides were filtered to 1% FDR using a decoy database strategy. A mininum score of 40 and differential score of 6 were required for modified peptides. Reporter ion quantification was used to extract intensities of the diagnostic HexNAc ions (138, 144, 168, 186, 204) from the spectra. O-GlcNAc peptides with a measured oxonium ion intensity ratio characteristic of GlcNAc (138/144 > 3) rather than GalNAc are indicated [
26,
27]. Reporter ion #6 was included to extract intensities for
m/
z 366 (HexNAc-Hex) to remove peptides with a higher level of glycosylation. Putative O-GlcNAcylated peptides were filtered to retain those with at least 3 HexNAc oxonium ions including 204
m/
z in HCD spectra. The probabilities of O-GlcNAc site assignment are reported.
2.9. Identification of O-GlcNAc Sites of GATAD2B
Immunoprecipitated, gel-purified GATAD2B was reduced with dithiothreitol, alkylated with iodoacetamide, and in-gel digested with trypsin. Extracted peptides were analyzed by LC-MS/MS as described above with the following modifications. HCD fragmentation was performed with a precursor isolation window of 1.7 m/z, 200% normalized AGC, a maximum injection time of 105 ms, and HCD collision energy of 40%. An HCD product ion of 204.0867 within the top 20 ions with a 15 ppm mass tolerance was used to trigger acquisition of an ETD scan. ETD fragmentation was performed using a 1.7 m/z isolation window, 15,000 resolution, 120 ms maximum injection time, 800% normalized AGC target, using the calibrated charge dependent ETD reaction parameters.
The raw files were searched using MaxQuant version 2.0.1.0 (Max Plank Institute) as described above against human GATAD2B downloaded from UniProt. All reported O-GlcNAc modified peptides of GATAD2B were manually verified. A previously identified site of O-GlcNAcylation on tyrosine within GATAD2B was not observed [
25]. HCD MS/MS spectra contained the diagnostic GlcNAc fragmentation pattern with complete neutral loss of the monosaccharide from the peptide precluding site assignment. Peptide fragmentation by ETD was insufficient for site assignment.
2.10. Detection of GATAD2B Protein Interactions
MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle’s Medium (Corning 10-014-CV) supplemented with 10% fetal bovine serum. Cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C and were passaged by trypsinization every two to three days. Cells at 90–95% confluency were treated with Thiamet G (Sigma-Aldrich) at a final concentration of 10 µM or OSMI-1 (Sigma-Aldrich) at a final concentration of 37 µM, or DMSO. After 24 h, cells were washed three times with cold PBS and lysed with cell lysis buffer (Cell Signaling #9803S) amended with universal nuclease (Thermo Scientific) and brought to a final concentration of 200 mM NaCl. Protein concentration was determined using BCA assay. One mg of protein was incubated with anti-GATAD2B antibody (Bethyl, Laboratories, Montgomery, TX, USA, cat#A301-281A) or rabbit IgG (Cell-Signaling, cat# 2729) as negative control overnight at 4 °C. Washed protein A/G Plus Agarose beads (Santa-Cruz, cat# sc2003) (30 uL) were added and protein complexed immunoprecipitated for 2 h in at 4 °C. Beads were washed wih 25 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP40, 5% Glycerol) five times and once with PBS. Proteins were eluted with 3× SDS sample loading buffer (60 uL) and denatured at 95 °C for 8 min. Proteins were separated by 4–20% glycine SDS gel and each lane was in-gel trypsin digested, cysteines were reduced and alkylated with iodoacetamide. Extracted peptides were separated and analyzed on an EASY nLC 1200 System (Thermo Scientific) in-line with the Orbitrap Exploris 480 (Thermo Scientific) with instrument control software v. 4.2.28.14. Two µg of peptides were pressure loaded onto a C18 reversed phase column (Acclaim PepMap RSLC, 75 µm × 25 cm (2 µm, 100 Å) ThermoFisher cat. # 164941) and separated using a gradient of 0–35% B in 90 min (Solvent A: 5% acetonitrile, 0.2% formic acid; Solvent B: 80% acetonitrile, 0.2% formic acid) at a flow rate of 300 nL/min.
Data were acquired in data-dependent acquisition (DDA) and data independent acquisition (DIA) modes to build a hybrid DDA and DIA library for database searching. High-resolution massspectometry analysis was carried out using FTMS with a survey scan of 60,000, in a mass range of m/z 375–1575. The mass spectra of the most abundant precursors were collected every 3 s. For the survey MS scan, automatic gain was set at 300%. For the MS/MS scan, automatic gainwas set at 100%. HCD fragmentation was carried out with 1.4 m/z precursor isolation window, 40 ms maximum injection time and 33% HCD collision energy. MS/MS data was collected at 15,000 resolution with peptide-specific monoisotopic-precursor selection. Resequencing for 20 s was carried out and precursors within 10 ppm mass tolerance were dynamically excluded. Advanced peak determination was enabled, and precursor ions with charged state of 1, >6 or undetermined were excluded. Mass spectra were also acquired in data-independent mode with a high resolution (60,000) FTMS survey scan, mass range of m/z 380–985, with an automatic gain control target value of 100% and a maximum injection time of 100 ms. The AGC target value for fragment spectra was set at 200%. 59 windows of 10 m/z scanning from 380–980 was used. The resolution was 15,000 and injection time 40 ms. HCD collision energy of 33%.
Data were searched using Spectronaut 17 using the Direct DIA+ algorithm (Biognosys, Newton, MA, USA) against a reviewed human protein database downloaded from Uniprot on (20 June 2022 with 20,386 entries). A decoy database was generated in Spectronaut and a contaminant database (downloaded with MSFragger) was included. Two peptides were required for protein identification. Data were uploaded into Perseus v1.6.15.0 (Max Planck Institute) for data processing and annotation with complexes from the Corum database (4.1 release November 2022), Reactome pathways (downloaded 03172023), Go terms and Uniprot gene names (downloaded from Uniprot 080520_SP_HUMAN_n20353). Binary comparisons were performed between GATAD2B IP vs IgG control; GATAD2B_ThiametG IP vs IgG control; and GATAD2B_OSMI IP vs IgG control. For each binary comparison, log2 transformed protein intensities were filtered to retain proteins observed in all 3 replicates of the GATAD2B IP. Missing values for the IgG controls were imputed with 0.3 width and 1.8 downshift from the total matrix. A paired two sample t-test was performed against control samples run on the same gel. The log2 fold change versus the –log10 p-values were plotted in volcano plots.
2.11. Co-Immunoprecipitation
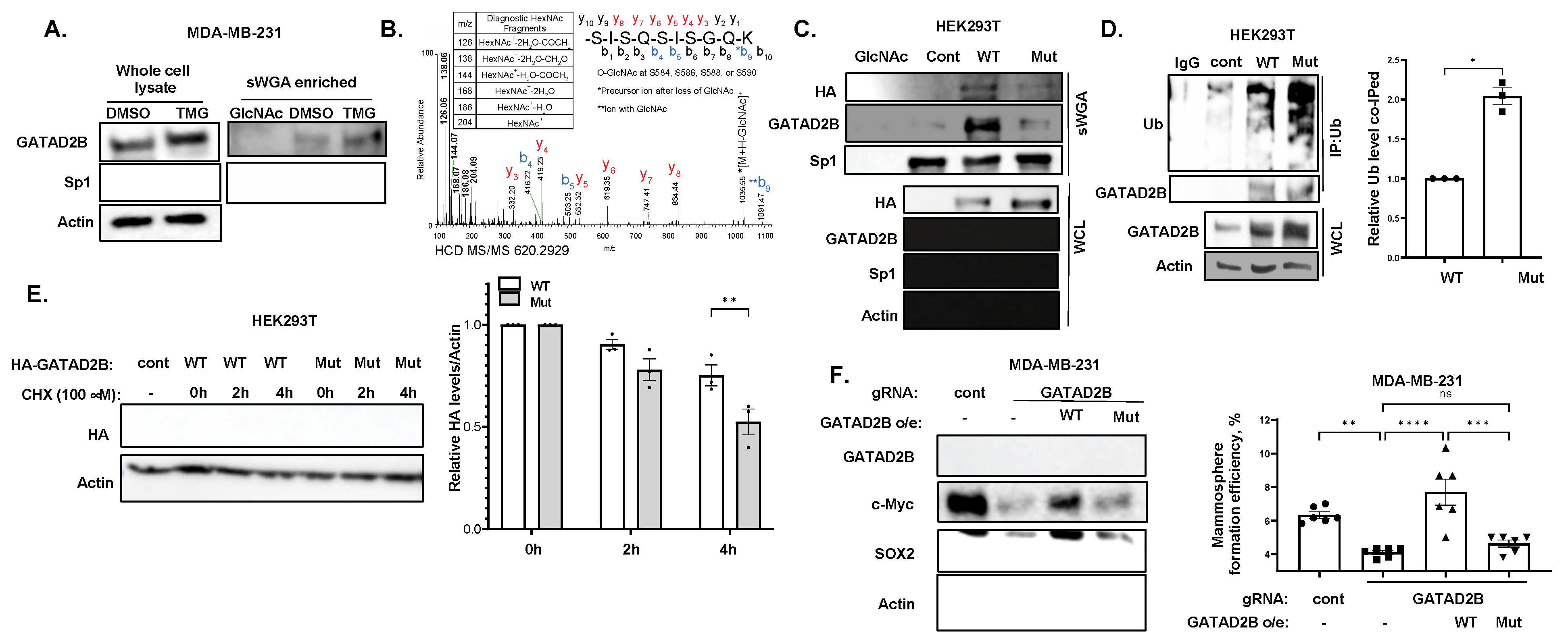
Cells were allowed to grow in monolayer and were treated with 100 µM of OGT inhibitor OSMi or with control of DMSO for 24 h. Cells were then harvested, washed and lysed in RIPA lysis buffer as described above. Bradford assay was used to quantify protein concentration. 1000 µg of protein was incubated with anti-GATAD2B (Bethyl, cat #A301-281A) and washed protein A/G Plus Agarose (Santa-Cruz, cat #sc2003) for overnight at 4 °C. Normal rabbit IgG (Cell-Signaling, cat #2729) was used as control. After incubation, samples were centrifuged and extensively washed with RIPA buffer before analyzing by Western blot as described above. Indicated targets were detected using the following specific antibodies: Anti-GATAD2B (Cell-Signaling, cat #73098), anti-ITCH (Cell-Signaling, cat #12117), anti-HA (Cell-Signaling, cat #3724), anti-Ubiquitin (Cell-Signaling, cat #3933).
To identify the Ubiquitination level of Wild-type and mutant GATAD2B, HEK-293T cells were transfected with plasmid encoding for GATAD2B-WT or GATAD2B-Mut. Cancer cells expressing GATAD2B-WT or GATAD2B-Mut were washed and lysed using RIPA buffer supplemented with protease inhibitors. 1000 µg from each sample were used for immunoprecipitation using anti-GATAD2B (Bethyl, cat #A301-281A) as described above. Immunoprecipitated proteins were resolved and analyzed by Western blot using the following indicated antibodies: Anti-GATAD2B (Cell-Signaling, cat #73098), anti-Ubiquitin (Cell-Signaling, cat #3933).
To confirm the role of ITCH in Ubiquitinating GATAD2B, breast cancer cells were infected with lentiviral vector delivering control or ITCH shRNA. Cells were then treated with OGT inhibitor OSMi (100 µM) for 24 h before being collected for cell lysate. 1000 µg from each sample were used for immunoprecipitation using anti-GATAD2B (Bethyl, cat #A301-281A) as described above. Immunoprecipitated proteins were analyzed by Western blot using the following indicated antibodies: Anti-GATAD2B (Cell-Signaling, cat #73098), anti-ITCH (Cell-Signaling, cat #12117), anti-Ubiquitin (Cell-Signaling, cat #3933).
2.12. Succinylated Wheat Germ Agglutinin Assay
Cells were allowed to grow in monolayer as described before and was treated with 2 µM of OGA inhibitor Thiamet-G (TMG) for 3 h to increase the level of total O-GlcNAc. After 3 h, cells were collected, and cell lysates were prepared as described above. 250 µg of protein from each sample was incubated with washed succinylated Wheat Germ Agglutinin Agarose (sWGA) beads (Vector Laboratories, cat #AL-1023S-5) for overnight at 4 °C. A sample with sWGA bead prebound to N-acetylglucosamine (GlcNAc) (Sigma, cat #A8625) was used as a negative control. After incubation, beads were pelleted and extensively washed before analyzing by Western blot as described above. The following specific antibodies were used to detect proteins of interest: Anti-GATAD2B (Cell-Signaling, cat #73098), anti-Sp1 (Cell-Signaling, cat #5931), anti-HA (Cell-Signaling, cat #3724).
2.13. Quantitative RT-PCR (qRT-PCR)
Breast cancer cells were collected for total RNA extraction using Trizol reagent (Invitrogen) as per manufacturer’s instruction. Briefly, Trizol reagent was added directly to monolayer of breast cancer cells. The mixture of cells and Trizol was transferred into a new tube, mixed with cloroform to extract total RNA. Isopropanol was added to the RNA fraction to precipitate RNA, which was subsequently washed with 70% ethanol. RNA pellet was allowed to air-dry and resuspended in nuclease-free water. RNA concentration was determined by measuring OD260 nm using NanoDrop. mRNA level of target genes was assessed by qRT-PCR using Brilliant II Master Mix Kit (Agilent) as per manufacturer’s instruction. qRT-PCR assay was conducted using qRT-PCR Applied Biosystem 7500 (Applied Biosystem, Waltham, MA, USA). The following Taqman probes were used to quantify mRNA level of targeted genes: PPIA (Hs04194521_s1), OCT4 (Hs04260367), SOX2 (Hs04234836_s1), c-MYC (Hs00905030_m1), Nanog (Hs02387400_g1) and GATAD2B (Hs00372672_m1).
2.14. Apoptosis Assay
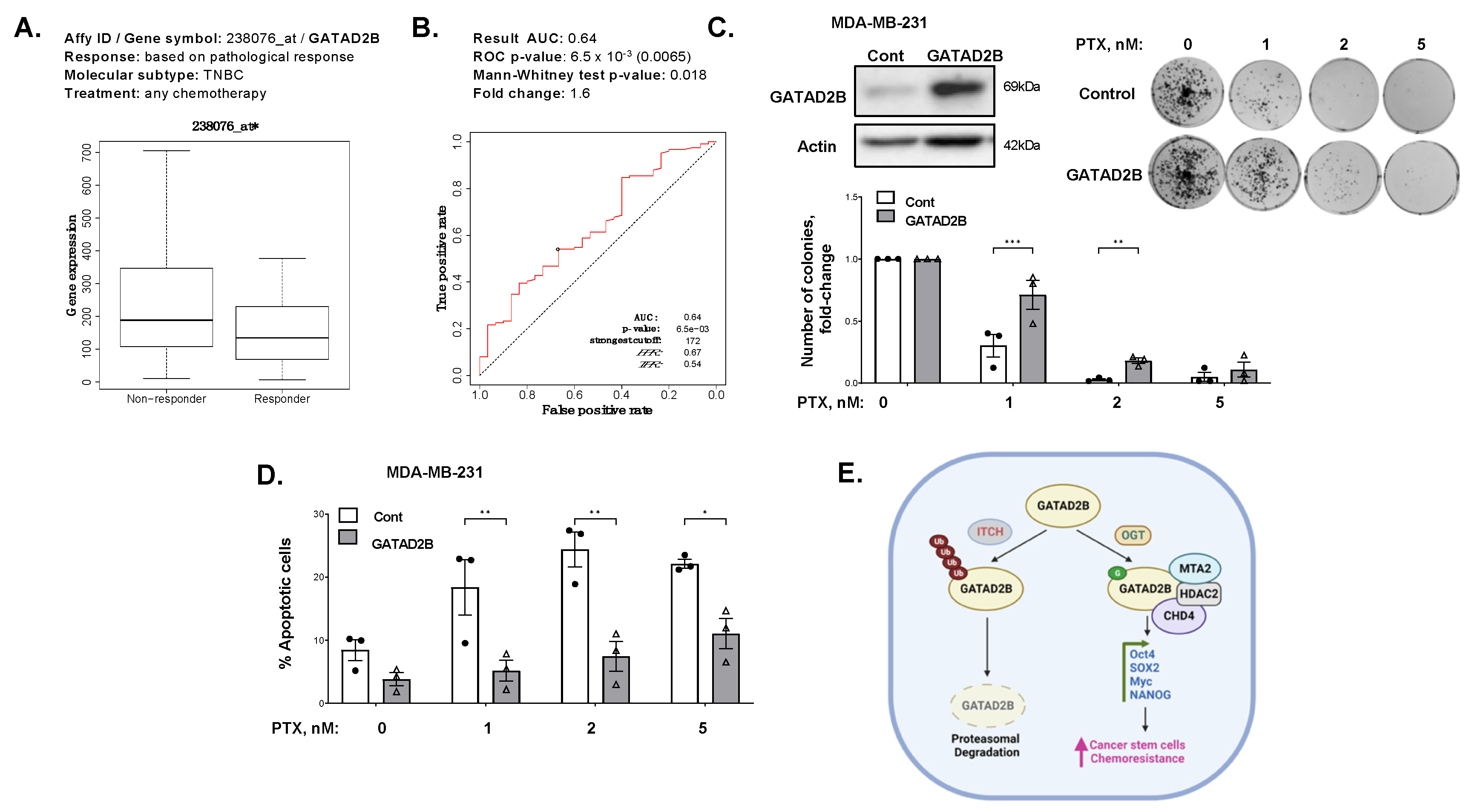
Breast cancer cells growing in monolayer were treated with increasing concentration of paclitaxel or with control of DMSO for 48 h. All cells were harvested and washed with PBS. Cells were analyzed using Annexin V-FITC Apoptosis detection kit (Biolegend, San Diego, CA, USA) as per manufacturer’s instruction. In brief, 106 cells were resuspended in 1 mL of 1× provided binding buffer to final concentration of 106 cells/mL. 100 μL of cell suspension was mixed with 5 μL of provided FITC-Annexin V and 5 μL of Propidium Iodide and incubated for 15 min in the dark at room temperature. After incubation, the volume of each sample was brought up to 500 μL using 1× binding buffer. Flow cytometry analysis was conducted using Guava flowcytometry system (MillporeSigma). Unstained and single stain samples were used to determine the basal fluorescence signal.
2.15. Clonogenic Survival Assay
Breast cancer cells growing in monolayer were treated with increasing dose of paclitaxel, or with control DMSO for 48 h. Cells were harvested and 1000 cells from each sample were plated and allowed to grow for 10–14 days to form colonies. Colonies were subsequently stained with crystal violet and counted. Counted number of colonies was normalized against control of DMSO treatment.
2.16. Database Analysis
OncoPrint plot of genes coding for NuRD complex was generated using available online database analysis platform cBioportal (
https://www.cbioportal.org accessed on 31 March 2023) using the Breast cancer METABRIC database (Invasive Breast Carcinoma: METABRIC, Nature 2012 and Nat Commun 2016). OncoPrints of GATAD2B, GATAD2A, CHD4, MTA2, HDAC2 and RBBP4 were analyzed. Correlation in expression of GATAD2B and OGT was analyzed using cBioportal platform, using the breast cancer METABRIC database. Co-expression of OGT mRNA was plotted against GATAD2B mRNA [
28,
29].
GATAD2B level in breast cancer subtypes was analyzed using available online database at UALCAN (
https://ualcan.path.uab.edu/analysis.html, accessed on 31 March 2023) using CPTAC database (Breast cancer, total-protein, major subclasses) [
30].
Kaplan-Meier survival plots were created using Kaplan-Meier plotter, an available online database analysis platform (
https://kmplot.com/analysis, accessed on 31 March 20233) [
31]. The relevant mRNA level of GATAD2B in basal-type breast cancer (auto select best cutoff, overall survival, PAM50 subtype: basal) was plotted against the overall survival of breast cancer patients. The relevant protein level of GATAD2B in all breast cancer (auto select best cutoff, overall survival, Tang_2018) was plotted against overall survival of breast cancer patients. The expression of GATAD2B and relevant ROC plot in breast cancer patients, which differentially responded to any chemotherapy in five years (relapse-free survival at 5 years, any chemotherapy), were generated using available online ROC plotter (
https://www.rocplot.org/site/treatment, accessed on 31 March 2023) [
32].
2.17. Statistical Analysis
Data from at least 3 biological repeats is shown as mean ± SEM. Statistical analysis was conducted using Graphpad Prism 9.0 and representative p-value was presented as * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
The development of breast cancer is heavily influenced by signals from the tumor microenvironment, including the nutrient status, which is sensed by nutrient sensing mechanisms to modulate cellular activities [
9]. One such mechanism is the hexosamine biosynthetic pathway, which utilizes glucose and other substrates from major metabolic pathways to produce UDP-GlcNAc [
9]. This nucleotide sugar serves as a substrate for extensive N-linked and O-linked glycosylation, or for O-GlcNAcylation of intracellular proteins [
9,
10,
12]. The process of O-GlcNAcylation and its enzyme OGT function to couple metabolism to signaling transduction in cancer cells [
12]. Increased OGT and O-GlcNAc are elevated in various cancers, including breast cancer. Elevated levels of OGT and O-GlcNAc in breast cancer promote breast tumor growth and metastasis [
46,
47]. Recent studies have shown that OGT and O-GlcNAc play a critical role in maintaining and driving tumor initiation and CSCs phenotypes. Elevated expression of OGT increases mammosphere formation, CSCs population and expression of CSCs markers [
17]. However, the mechanism of how OGT and O-GlcNAc control CSCs maintenance and function is still unclear. Here, using a global proteomic analysis, we identified GATAD2B of the NuRD complex as a target of OGT and O-GlcNAc in breast cancer. The expression of GATAD2B is critical and sufficient to maintain and promote CSCs phenotype of breast cancer cells. GATAD2B also regulates expression of CSCs markers, including Oct4, SOX2, c-Myc and NANOG, at both the mRNA and protein level. We also confirmed that GATAD2B is a critical downstream effector of OGT and O-GlcNAc in controlling the CSCs properties. In this mechanism, direct O-GlcNAcylation of GATAD2B protects from ubiquitination and proteasomal degradation, thus enhancing the protein stability. In addition, we identify ITCH as a E3 ligase that interacts with GATAD2B and regulates it degradation. This interaction between GATAD2B and ITCH is enhanced when the enzymatic activity of OGT is inhibited. ITCH knockdown increased the level of GATAD2B, but also elevated the mammosphere formation of breast cancer cells and increased cancer stem cell factors SOX2 and c-Myc. Importantly, genetic inhibition of ITCH was also able to rescue the level of GATAD2B and mammosphere formation in the presence of OGT inhibitor, which further indicated the critical role of OGT in regulating GATAD2B level to drive CSCs function. Moreover, we for the first time showed that O-GlcNAcylation within the C-terminus of GATAD2B is critical for GATAD2B stability, as well as for the CSCs properties of breast cancer cells. Other groups have shown that GATAD2B can be O-GlcNAcylated at other sites [
48]. However, the functional role of these other GATAD2B O-GlcNAcylation sites need to be furthered examined. Collectively, we showed a novel regulatory mechanism of GATAD2B by OGT, which is pivotal for CSCs maintenance and function in breast cancer (
Figure 7E).
GATAD2B is a component of the NuRD complex, and functions as a scaffolding protein for NuRD complex assembly [
49]. Dysregulation of GATAD2B was reported in multiple neurological disorders, which is potentially linked to the role of GATAD2B and NuRD complex in regulating function of stem cells and progenitor cells [
50,
51]. In addition, our study shows that GATAD2B is highly upregulated in breast cancer cells and in patient samples. However, the role of GATAD2B in breast cancer has not yet been reported. We for the first time showed that GATAD2B is critical for CSCs function in breast cancer and is regulated directly by OGT via O-GlcNAcylation. This role of GATAD2B in CSCs aligns with the reported role of GATAD2B in promoting metastasis in KRAS-mutant lung cancer, strengthening the role of GATAD2B in cancer [
21]. The regulation GATAD2B O-GlcNAcylation possibly results in changes in NuRD complex formation, which subsequently controls gene expression by remodeling chromatin. Although GATAD2B has an analog, GATAD2A in the NuRD complex, recent evidence suggests that NuRD complexes containing GATAD2B as opposed to GATAD2A may possess distinct functions [
20,
50,
52]. Consistent with this idea, our results show that GATAD2B, but not GATAD2A, can regulate CSCs function in breast cancer further implicating a specific role of GATAD2B in CSCs. It will be interesting to identify in the future the specific NuRD complex bound to GATAD2B that regulates CSC functions.
O-GlcNAcylation of target protein alters the protein stability, localization, and protein-protein interaction, thus facilitating the modulation of protein functions [
12]. O-GlcNAcylation on GATAD2B increases the half-life of the NuRD complex component by impairing the interaction between GATAD2B and its E3-ligase, ITCH. ITCH promotes GATAD2B ubiquitination and subsequently its proteasomal degradation. Genetic inhibition of ITCH enhances the stability of GATAD2B, mammosphere formation and expression of CSCs factors in breast cancer cells. Importantly, inhibition of OGT enzymatic activity enhances GATAD2B and ITCH interaction reducing GATAD2B level and mammosphere formation. This reduction in GATAD2B and mammosphere formation is reversed by ITCH knockdown, implicating the importance of O-GlcNAcylation on GATAD2B stability and its function in promoting CSCs. Similarly, reducing O-GlcNAcylation on GATAD2B by mutating potential modification Serines to Alanines also shortens GATAD2B half-life and increases ubiquitination level of GATAD2B, subsequently impairs mammosphere formation of breast cancer cells. This finding, for the first time, points to the novel regulating mechanism of OGT on GATAD2B via modulating GATAD2B degradation in an ITCH-dependent manner.
The E3-ligase ITCH plays a crucial role during the development of breast tumors. In breast cancer, inhibited expression of ITCH led to an increase in the activity of the Wnt/β-catenin signaling promoting the growth of breast cancer in vitro and in vivo [
53]. In contrast, increased expression of ITCH resulted in decreased signaling activity of the Wnt/β-catenin, reduced cell proliferation and tumor growth [
53]. In stem cells, ITCH negatively regulates the function and homeostasis of hemopoietic stem cells, where reduced expression of ITCH led to increased expression of pro-self-renewing factor Notch1, increased proliferation and sustained function of progenitor cells [
54]. In addition, ITCH functions as an E3-ligase of Oct4, a key transcription factor for maintaining self-renewal, to contribute to regulate the fate of embryonic stem cells [
55]. However, the potential role of ITCH in regulating cancer stem-like cells properties has not been reported. Here, we showed that genetic inhibition of ITCH increased expression of GATAD2B, SOX2 and MYC, as well as mammosphere formation and CSCs population detected by ALDEFLOUR in multiple breast cancer cell lines. The function of ITCH in regulating CSCs in breast cancer could be, in part, a result of ITCH regulation on GATAD2B stability. Together, these data show a crucial role of ITCH in mediating GATAD2B levels and regulating CSCs in breast cancer. One important characteristic of CSCs is the potential in promoting resistance to anti-cancer therapies [
44]. Overexpression of GATAD2B increases the CSCs properties in breast cancer cells, but also promotes resistance to apoptosis induced by paclitaxel in vitro. Increased expression of GATAD2B in breast cancer cells protects from paclitaxel-mediated apoptosis and clonogenic survival and requires O-GlcNAcylation of GATAD2B. These findings, together with high expression of GATAD2B in breast cancer patients that are non-responsive to chemotherapy, suggests the potential of GATAD2B to serve as a clinical prognosis marker for breast cancer patients.



 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}