
Sex Disparity in Cancer: Role of Autophagy and Estrogen Receptors



Abstract
1. Introduction
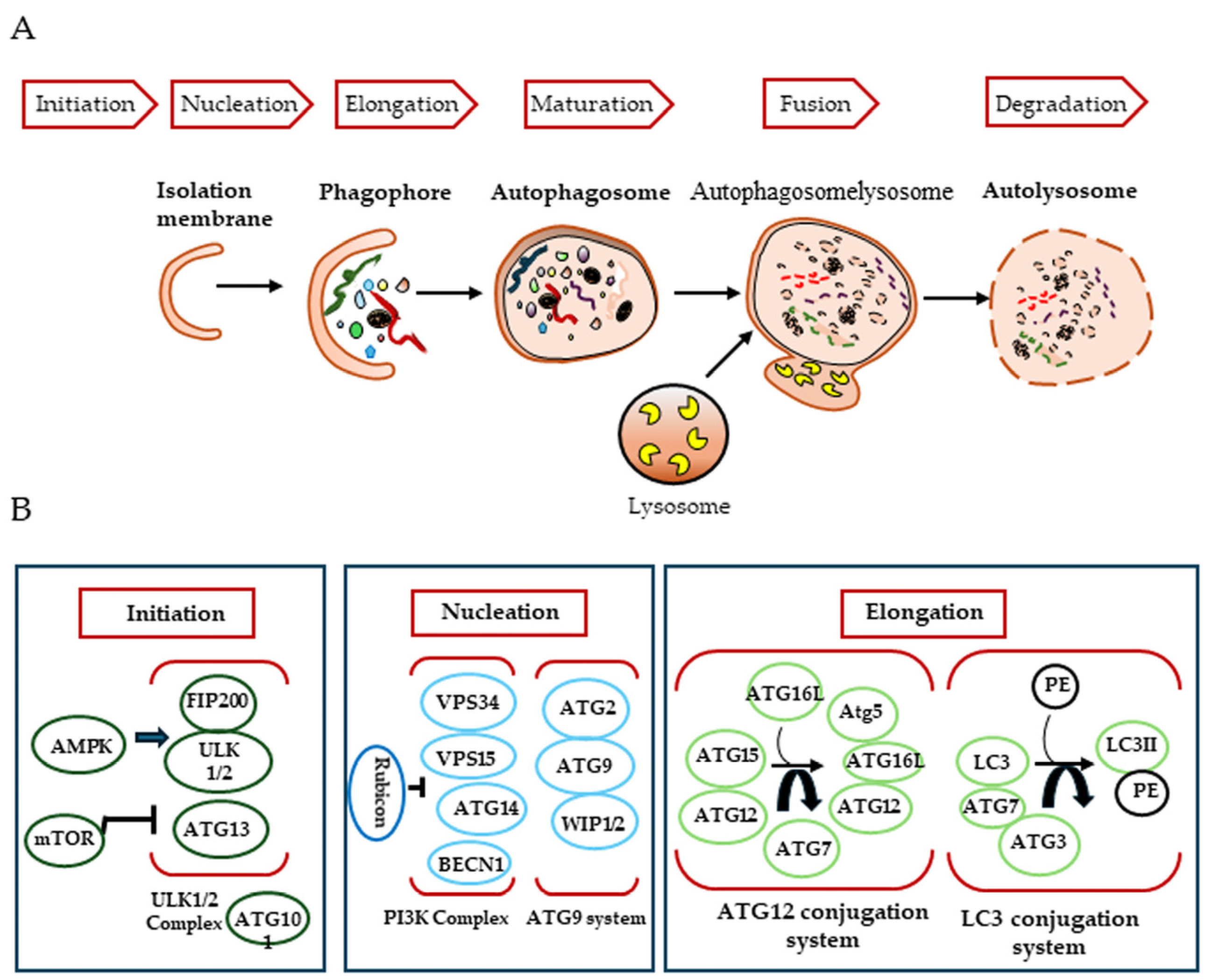
2. Autophagy
2.1. Biogenesis of the Autophagosome
2.2. Key Regulators of the Autophagic Process
3. Autophagy and Cancer
4. Sex Differences in Autophagy
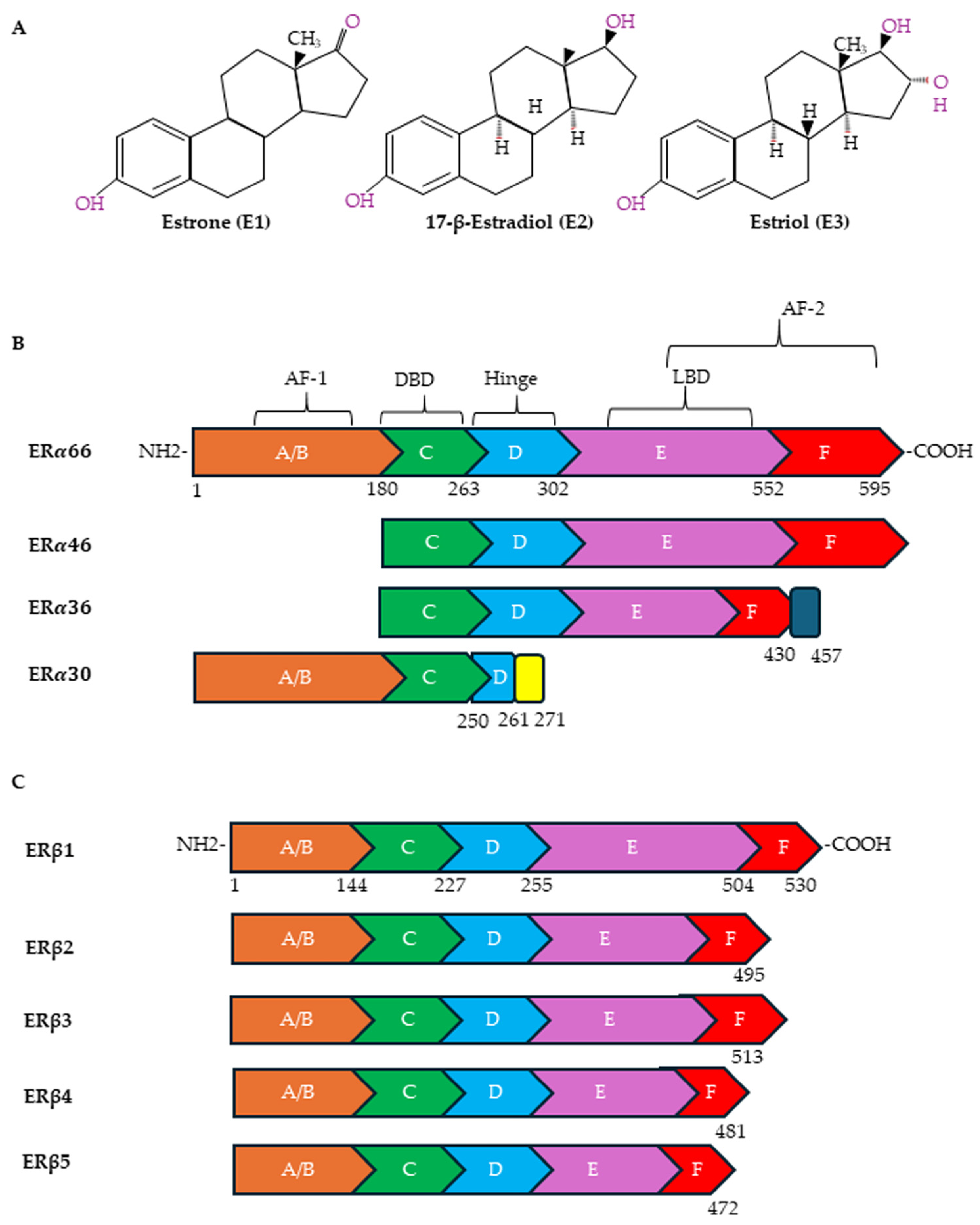
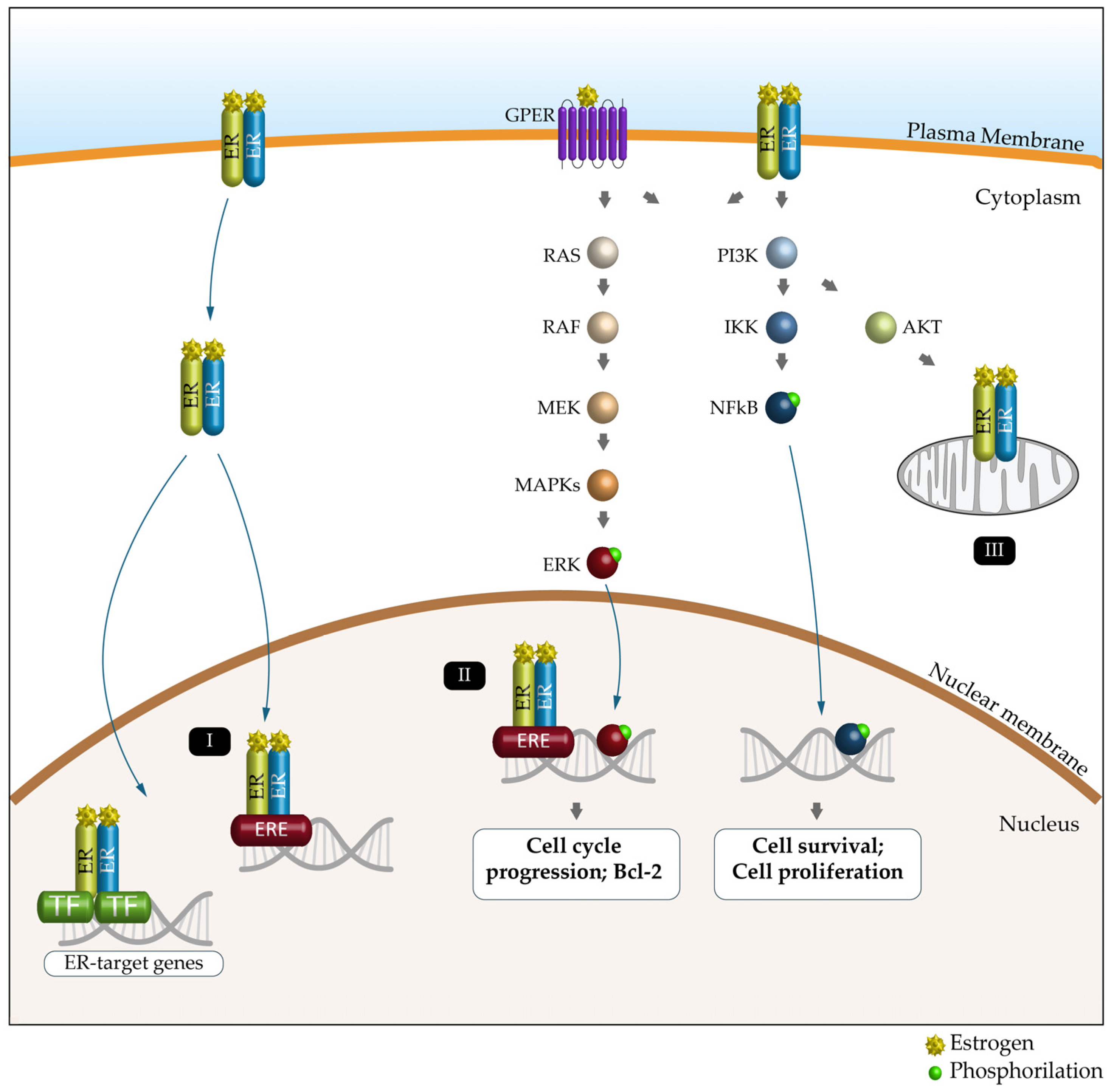
5. Estrogen Receptors and Cancer
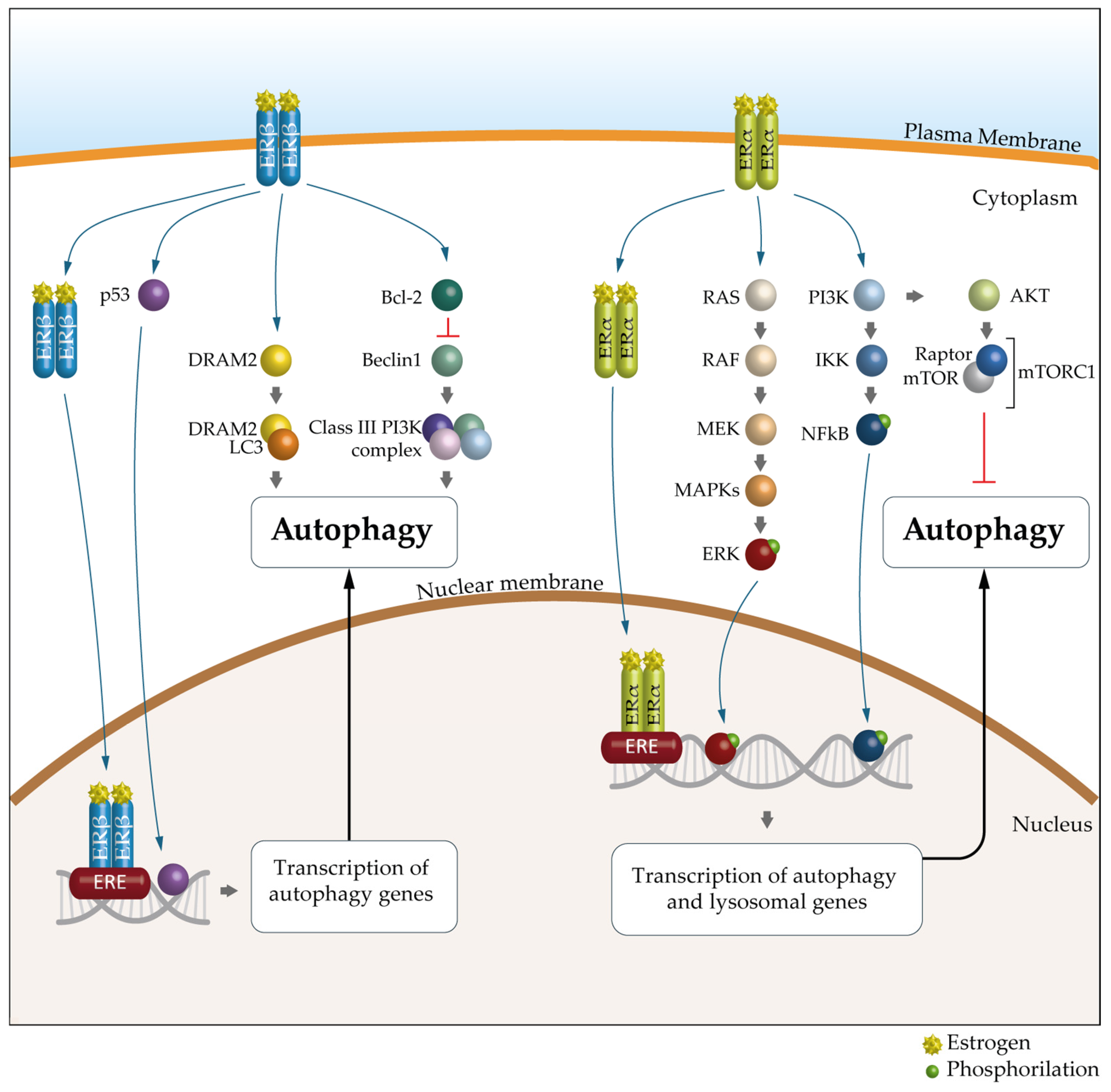
6. Autophagy and Estrogen Receptors
6.1. Colorectal Cancer (CRC)
6.2. Gastric Cancer (GC)
6.3. Non-Small Cell Lung Carcinoma (NSCLC)
6.4. Glioblastoma (GBM)
6.5. Hematological Malignancies (HMs)
7. ERs and Autophagy as Potential Targets in Antitumor Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagy in the eukaryotic cell. Eukaryot. Cell 2002, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Clayton, J.A. Studying both sexes: A guiding principle for biomedicine. FASEB J. 2016, 30, 519–524. [Google Scholar] [CrossRef]
- Stachenfeld, N.S.; Mazure, C.M. Precision medicine requires understanding how both sex and gender influence health. Cell 2022, 185, 1619–1622. [Google Scholar] [CrossRef]
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; et al. Sex differences in cancer mechanisms. Biol. Sex Differ. 2020, 11, 17. [Google Scholar] [CrossRef]
- Word Health Organization. Available online: https://www.who.int/health-topics/#G (accessed on 6 February 2025).
- Pape, M.; Miyagi, M.; Ritz, S.A.; Boulicault, M.; Richardson, S.S.; Maney, D.L. Sex contextualism in laboratory research: Enhancing rigor and precision in the study of sex-related variables. Cell 2024, 187, 1316–1326. [Google Scholar] [CrossRef]
- Rubin, J.B.; Abou-Antoun, T.; Ippolito, J.E.; Llaci, L.; Marquez, C.T.; Wong, J.P.; Yang, L. Epigenetic developmental mechanisms underlying sex differences in cancer. J. Clin. Investig. 2024, 134, e180071. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Ann. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–4676. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Rieber, M.; Rieber, M.S. Sensitization to radiation-induced DNA damage accelerates loss of bcl-2 and increases apoptosis and autophagy. Cancer Biol. Ther. 2008, 7, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Roy, S.; Debnath, J. Autophagy and tumorigenesis. Semin. Immunopathol. 2010, 32, 383–396. [Google Scholar] [CrossRef]
- Boretto, C.; Actis, C.; Faris, P.; Cordero, F.; Beccuti, M.; Ferrero, G.; Muzio, G.; Moccia, F.; Autelli, R. Tamoxifen Activates Transcription Factor EB and Triggers Protective Autophagy in Breast Cancer Cells by Inducing Lysosomal Calcium Release: A Gateway to the Onset of Endocrine Resistance. Int. J. Mol. Sci. 2023, 25, 458. [Google Scholar] [CrossRef]
- Qi, H.; Jiang, Z.; Wang, C.; Yang, Y.; Li, L.; He, H.; Yu, Z. Sensitization of tamoxifen-resistant breast cancer cells by Z-ligustilide through inhibiting autophagy and accumulating DNA damages. Oncotarget 2017, 8, 29300–29317. [Google Scholar] [CrossRef]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2011, 11, 127–137. [Google Scholar] [CrossRef]
- Lorin, S.M.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Inoue, D.; Suzuki, T.; Mitsuishi, Y.; Miki, Y.M.; Suzuki, S.; Sugawara, S.; Watanabe, M.; Sakurada, A.; Endo, C.; Uruno, A.; et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012, 103, 760–766. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Liu, J.; Debnath, J. The evolving, multifaceted roles of autophagy in cancer. Adv. Cancer Res. 2016, 130, 1–53. [Google Scholar]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Alizadeh, J.; Kavoosi, M.; Singh, N.; Lorzadeh, S.; Ravandi, A.; Kidane, B.; Ahmed, N.; Mraiche, F.; Mowat, M.R.; Ghavami, S. Regulation of autophagy by carbohydrate and lipid metabolism in cancer. Cancers 2023, 15, 2195. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay between lipid metabolism and autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Sukocheva, O.A.; Furuya, H.; Ng, M.L.; Friedemann, M.; Menschikowski, M.; Tarasov, V.V.; Chubarev, V.N.; Klochkov, S.G.; Nneganova, M.E.; Mangoni, A.A.; et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol. Ther. 2020, 207, 107464. [Google Scholar] [CrossRef]
- Sukocheva, O.A. Expansion of Sphingosine Kinase and Sphingosine-1-Phosphate Receptor Function in Normal and Cancer Cells: From Membrane Restructuring to Mediation of Estrogen Signaling and Stem Cell Programming. Int. J. Mol. Sci. 2018, 19, 420. [Google Scholar] [CrossRef]
- Li, F.; Zhang, Y.; Lin, Z.; Yan, L.; Liu, Q.; Li, Y.; Pei, X.; Feng, Y.; Han, X.; Yang, J.; et al. Targeting SPHK1/S1PR3-regulated S-1-P metabolic disorder triggers autophagic cell death in pulmonary lymphangiomyomatosis (LAM). Cell Death Dis. 2022, 13, 1065. [Google Scholar] [CrossRef]
- Singh, M.K.; Han, S.; Kim, S.; Kang, I. Targeting Lipid Metabolism in Cancer Stem Cells for Anticancer Treatment. Int. J. Mol. Sci. 2024, 25, 11185. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Malik, F. Understanding the cell survival mechanism of anoikis-resistant cancer cells during different steps of metastasis. Clin. Exp. Metastasis 2022, 39, 715–726. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M.P. Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin. Oncol. 2014, 41, 195–216. [Google Scholar] [CrossRef]
- Antoon, R.; Overdevest, N.; Saleh, A.H.; Keating, A. Mesenchymal stromal cells as cancer promoters. Oncogene 2024, 43, 3545–3555. [Google Scholar] [CrossRef]
- Lista, P.; Straface, E.; Brunelleschi, S.; Franconi, F.; Malorni, W. On the role of autophagy in human diseases: A gender perspective. J. Cell. Mol. Med. 2011, 15, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving neurons show sex difference in autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E. Sex differences in autophagy contribute to female vulnerability in Alzheimer’s disease. Front. Neurosci. 2018, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Wang, L.; Klionsky, D.J.; Cheng, H.; Zhou, R. Sex differences in autophagy-mediated diseases: Toward precision medicine. Autophagy 2021, 17, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Itoh, T. Direct link between Atg protein and small GTPase Rab: atg16L functions as a potential Rab33 effector in mammals. Autophagy 2008, 4, 824–826. [Google Scholar] [CrossRef]
- Yang, M.; Liang, C.; Swaminathan, K.; Herrlinger, S.; Lai, F.; Shiekhattar, R.; Chen, J.F. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016, 2, e1601167. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef]
- Dunford, A.; Weinstock, D.M.; Savova, V.; Schumacher, S.E.; Cleary, J.P.; Yoda, A.; Sullivan, T.J.; Hess, J.M.; Gimelbrant, A.; Beroukhim, R.; et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 2017, 49, 10–16. [Google Scholar] [CrossRef]
- Ruggieri, A.; Barbati, C.; Malorni, W. Cellular and molecular mechanisms involved in hepatocellular carcinoma gender disparity. Int. J. Cancer 2010, 127, 499–504. [Google Scholar] [CrossRef]
- Shi, Y.; Han, J.J.; Tennakoon, J.B.; Mehta, F.F.; Merchant, F.A.; Burns, A.R.; Howe, M.K.; McDonnell, D.P.; Frigo, D.E. Androgens promote prostate cancer cell growth through induction of autophagy. Mol. Endocrinol. 2013, 27, 280–295. [Google Scholar] [CrossRef]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Khramtsov, A.; Tretiakova, M.S.; Krausz, T.N.; et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breastcancer progression. Mol. Syst. Biol. 2008, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.W.; Yang, S.H.; Sarkar, S.N.; Pearce, V. Estrogen actions on mitochondria—Physiological and pathological implications. Mol. Cell Endocrinol. 2008, 290, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Maselli, A.; Pierdominici, M.; Vitale, C.; Ortona, E. Membrane lipid rafts and estrogenic signaling: A functional role in the modulation of cell homeostasis. Apoptosis 2015, 20, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef]
- Thiebaut, C.; Konan, H.P.; Guerquin, M.J.; Chesnel, A.; Livera, G.; Le Romancer, M.; Dumond, H. The role of ERα36 in development and tumor malignancy. Int. J. Mol. Sci. 2020, 21, 4116. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for estrogen receptor alpha36 in cancer progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Zhu, H.; Huang, Y.; Su, H.; Ma, Y.; Tao, Y.; Liao, D.J.; Liu, Y.; Feng, Z. Identification of a novel human estrogen receptor-α splice variant able to enhance malignant biological behaviors of breast cancer cells. Oncol. Lett. 2018, 15, 5339–5344. [Google Scholar] [CrossRef]
- Yan, S.; Wang, J.; Chen, H.; Zhang, D.; Imam, M. Divergent features of ERβ isoforms in triple negative breast cancer: Progress and implications for further research. Front. Cell Dev. Biol. 2023, 11, 1240386. [Google Scholar] [CrossRef]
- Cvoro, A.; Tzagarakis-Foster, C.; Tatomer, D.; Paruthiyil, S.; Fox, M.S.; Leitman, D.C. Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol. Cell 2006, 21, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Karas, R.H. Emerging evidence of the importance of rapid, non-nuclear estrogen receptor signaling in the cardiovascular system. Steroids 2013, 78, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Cheng, Z. Hormonal regulation of metabolism-recent lessons learned from insulin and estrogen. Clin. Sci. 2023, 137, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Sui, C.Y.; Chen, Q.; Chen, X.P.; Zhang, H.; Zhou, X.P. Upregulation of cell surface estrogen receptor alpha is associated with the mitogen-activated protein kinase/extracellular signal-regulated kinase activity and promotes autophagy maturation. Int. J. Clin. Exp. Pathol. 2015, 8, 8832–8841. [Google Scholar]
- Noh, B.; McCullough, L.D.; Moruno-Manchon, J.F. Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome. Neural Regen. Res. 2023, 18, 31–37. [Google Scholar]
- Le, T.Y.; Ashton, A.W.; Mardini, M.; Stanton, P.G.; Funder, J.W.; Handelsman, D.J.; Mihailidou, A.S. Role of androgens in sex differences in cardiac damage during myocardial infarction. Endocrinology 2014, 155, 568–575. [Google Scholar] [CrossRef]
- Turei, D.; Foldvari-Nagy, L.; Fazekas, D.; Fazekas, D.; Módos, D.; Kubisch, J.; Kadlecsik, T.; Demeter, A.; Lenti, K.; Csermely, P.; et al. Autophagy regulatory network—A systems-level bioinformatics resource for studying the mechanism and regulation of autophagy. Autophagy 2015, 11, 155–165. [Google Scholar] [CrossRef]
- Dong, L.; Wang, W.; Wang, F.; Stoner, M.; Reed, J.C.; Samudio, I.; Kladde, M.P.; Vyhlidal, C.; Safe, S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J. Biol. Chem. 1999, 274, 32099–32107. [Google Scholar] [CrossRef]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Cianfanelli, V.; Salvioli, R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 2013, 9, 241–243. [Google Scholar] [CrossRef]
- Zhou, F.; Jin, J.; Zhou, L.; Wu, L.; Cao, Y.; Yan, H.; Huang, Q.; Wang, L.; Zou, X. Suppression of estrogen receptor-beta promotes gastric cancer cell apoptosis with induction of autophagy. Am. J. Transl. Res. 2020, 12, 4397–4409. [Google Scholar]
- Pierdominici, M.; Maselli, A.; Locatelli, S.L.; Ciarlo, L.; Careddu, G.; Patrizio, M.; Ascione, B.; Tinari, A.; Carlo-Stella, C.; Malorni, W.; et al. Estrogen receptor β ligation inhibits Hodgkin lymphoma growth by inducing autophagy. Oncotarget 2017, 8, 8522–8535. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Dai, G.; Li, Y.; Xu, L.; Liu, G. Intricate roles of estrogen and estrogen receptors in digestive system cancers: A systematic review. Cancer Biol. Med. 2024, 21, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Abancens, M.; Bustos, V.; Harvey HMcBryan, J.; Harvey, B.J. Sexual dimorphism in colon cancer. Front. Oncol. 2020, 10, 607909. [Google Scholar] [CrossRef]
- Benedix, F.; Kube, R.; Meyer, F.; Schmidt, U.; Gastinger, I.; Lippert, H. Comparison of 17,641 patients with right- and left-sided colon cancer: Differences in epidemiology, perioperative course, histology, and survival. Dis. Colon Rectum 2010, 53, 57–64. [Google Scholar] [CrossRef]
- Slattery, M.L.; Potter, J.D.; Curtin, K.; Edwards, S.; Ma, K.N.; Anderson, K.; Schaffer, D.; Samowitz, W.S. Estrogens reduce, and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res. 2001, 61, 126–130. [Google Scholar]
- Nüssler, N.C.; Reinbacher, K.; Shanny, N.; Schirmeier, A.; Glanemann, M.; Neuhas, P.; Nussler, A.K.; Kirschner, M. Sex-specific differences in the expression levels of estrogen receptor subtypes in colorectal cancer. Gend. Med. 2008, 5, 209–217. [Google Scholar] [CrossRef]
- Stevanato Filho, P.R.; Aguiar, S., Jr.; Begnami, M.D.; de Oliveira Ferreira, F.; Nakagawa, W.T.; Spencer, R.M.S.B.; Bezerra, T.S.; Boggiss, P.E.; Lopes, A. Estrogen receptor β as a prognostic marker of tumor progression in colorectal cancer with familial adenomatous polyposis and sporadic polyps. Pathol. Oncol. Res. 2018, 24, 533–540. [Google Scholar] [CrossRef]
- Rudolph, A.; Toth, C.; Hoffmeister, M.; Roth, W.; Herpel, E.; Schirmacher, P.; Chang-Claude, J. Colorectal cancer risk associated with hormone use varies by expression of estrogen receptor-β. Cancer Res. 2013, 73, 3306–3315. [Google Scholar] [CrossRef]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen receptors and their implications in colorectal carcinogenesis. Front. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef]
- Liang, R.; Lin, Y.; Yuan, C.L.; Liu, Z.H.; Li, Y.Q.; Luo, X.L.; Ye, J.Z.; Ye, H.H. High expression of estrogen-related receptor α is significantly associated with poor prognosis in patients with colorectal cancer. Oncol. Lett. 2018, 15, 5933–5939. [Google Scholar] [CrossRef]
- Ye, S.B.; Cheng, Y.K.; Zhang, L.; Wang, X.P.; Wang, L.; Lan, P. Prognostic value of estrogen receptor-α and progesterone receptor in curatively resected colorectal cancer: A retrospective analysis with independent validations. BMC Cancer 2019, 19, 933. [Google Scholar] [CrossRef] [PubMed]
- Ditonno, I.; Losurdo, G.; Rendina, M.; Pricci, M.; Girardi, B.; Ierardi, E.; Di Leo, A. Estrogen receptors in colorectal cancer: Facts, novelties and perspectives. Curr. Oncol. 2021, 28, 4256–4263. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Saha, J.; Pillai, S.; Lam, A.K.; Gopalan, V.; Islam, F. Implications of estrogen and its receptors in colorectal carcinoma. Cancer Med. 2023, 12, 4367–4379. [Google Scholar] [CrossRef]
- Luceri, C.; Femia, A.P.; Tortora, K.; D’Ambrosio, M.; Fabbri, S.; Fazi, M.; Caderni, G. Supplementation with phytoestrogens and insoluble fibers reduces intestinal carcinogenesis and restores ER-β expression in Apc-driven colorectal carcinogenesis. Eur. J. Cancer Prev. 2020, 29, 27–35. [Google Scholar] [CrossRef]
- Sun, W.; Lei, Y.; Jiang, Z.; Wang, K.; Liu, H.; Xu, T. BPA and low-Se exacerbate apoptosis and mitophagy in chicken pancreatic cells by regulating the PTEN/PI3K/AKT/mTOR pathway. J. Adv. Res. 2025, 67, 61–69. [Google Scholar] [CrossRef]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef]
- Jang, Y.C.; Leung, C.Y.; Huang, H.L. Association of hormone replacement therapy with risk of gastric cancer: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 12997. [Google Scholar] [CrossRef]
- Xu, C.Y.; Guo, J.L.; Jiang, Z.N.; Xie, S.D.; Shen, J.G.; Shen, J.Y.; Wang, L.B. Prognostic role of estrogen receptor alpha and estrogen receptor beta in gastric cancer. Ann. Surg. Oncol. 2010, 17, 2503–2509. [Google Scholar] [CrossRef]
- Zhou, J.; Teng, R.; Xu, C.; Wang, Q.; Guo, J.; Xu, C.; Li, Z.; Xie, S.; Shen, J.; Wang, L. Overexpression of ERα inhibits proliferation and invasion of MKN28 gastric cancer cells by suppressing β-catenin. Oncol. Rep. 2013, 30, 1622–1630. [Google Scholar] [CrossRef]
- Gan, L.; He, J.; Zhang, X.; Zhang, Y.J.; Yu, G.Z.; Chen, Y.; Pan, J.; Wang, J.J.; Wang, X. Expression profile and prognostic role of sex hormone receptors in gastric cancer. BMC Cancer 2012, 12, 566. [Google Scholar] [CrossRef]
- Wang, M.; Pan, J.Y.; Song, G.R.; Chen, H.B.; An, L.J.; Qu, S.X. Altered expression of estrogen receptor alpha and beta in advanced gastric adenocarcinoma: Correlation with prothymosin alpha and clinicopathological parameters. Eur. J. Surg. Oncol. 2007, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yuan, C.; Jin, Y.; Huang, H.; Sheng, X.; Wei, W.; Huang, X.; Li, L.; Lv, K.; Qiu, Z.; et al. ER-α36 mediates gastric cancer cell invasion. Int. J. Clin. Exp. Pathol. 2020, 13, 1550–1559. [Google Scholar] [PubMed]
- Tsai, C.H.; Kung, P.T.; Kuo, W.Y.; Tsai, W.C. Effect of time interval from diagnosis to treatment for non-small cell lung cancer on survival: A national cohort study in Taiwan. BMJ Open 2020, 10, e034351. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lara, V.; Avila-Costa, M.R. An overview of lung cancer in women and the impact of estrogen in lung carcinogenesis and lung cancer treatment. Front. Med. 2021, 8, 600121. [Google Scholar] [CrossRef]
- Almotlak, A.A.; Farooqui, M.; Siegfried, J.M. Inhibiting pathways predicted from a steroid hormone gene signature yields synergistic antitumor effects in NSCLC. J. Thorac. Oncol. 2020, 15, 62–79. [Google Scholar] [CrossRef]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef]
- Słowikowski, B.K.; Lianeri, M.; Jagodziński, P.P. Exploring estrogenic activity in lung cancer. Mol. Biol. Rep. 2017, 44, 35–50. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, L.; Wang, H.; Li, J.; Ruan, X.; Sun, B.; Zhi, J.; Zheng, X.; Gu, L.; Gao, M.; et al. FATS regulates polyamine biosynthesis by promoting ODC degradation in an ERβ-dependent manner in non-small-cell lung cancer. Cell Death Dis. 2020, 11, 839. [Google Scholar] [CrossRef]
- Pelejanou, V.; Anastasiou, E.; Bakogeorgou, E.; Notas, G.; Kampa, M.; Garcia-Milian, R.; Lavredaki, K.; Mousou, E.; Chinari, G.; Arapantoni, P.; et al. Estrogen receptor-alpha isoforms are the main estrogen receptors expressed in non-small cell lung carcinoma. Steroids 2019, 142, 65–76. [Google Scholar] [CrossRef]
- Márquez-Garbán, D.C.; Chen, H.W.; Fishbein, M.C.; Goodglick, L.; Pietras, R.J. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 2007, 72, 135–143. [Google Scholar] [CrossRef]
- Castellanos, M.R.; Fanous, E.; Thaker, R.; Flory, M.J.; Seetharamu, N.; Dhar, M.; Starr, A.; Strange, T.J. Expression patterns and clinical significance of estrogen receptor in non-small cell lung cancer. Pathol. Res. Pract. 2023, 241, 154298. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wu, R.; Jiang, Y.; Qiu, Z.; Chen, W.; Li, W. Overexpression of estrogen receptor beta is a prognostic marker in non-small cell lung cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 8686–8697. [Google Scholar] [PubMed]
- Ma, L.; Zhan, P.; Liu, Y.; Zhou, Z.; Zhu, Q.; Miu, Y.; Wang, X.; Jin, J.; Li, Q.; Lv, T.; et al. Prognostic value of the expression of estrogen receptor beta in patients with non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2016, 5, 202–207. [Google Scholar] [CrossRef]
- Mauro, L.V.; Dalurzo, M.; Carlini, M.J.; Smith, D.; Nunez, M.; Simian, M.; Lastiri, J.; Vasallo, B.; Bal de Kier Joffé, E.; Pallotta, M.G.; et al. Estrogen receptor β and epidermal growth factor receptor as early-stage prognostic biomarkers of non-small cell lung cancer. Oncol. Rep. 2010, 24, 1331–1338. [Google Scholar]
- Shimizu, K.; Hirami, Y.; Saisho, S.; Yukawa, T.; Maeda, A.; Yasuda, K.; Nakata, M. Membrane-bound estrogen receptor-α expression and epidermal growth factor receptor mutation are associated with a poor prognosis in lung adenocarcinoma patients. World J. Surg. Oncol. 2012, 10, 141. [Google Scholar] [CrossRef]
- Kawai, H.; Ishii, A.; Washiya, K.; Konno, T.; Kon, H.; Yamaya, C.; Ono, I.; Minamiya, Y.; Ogawa, J. Estrogen receptor alpha and beta are prognostic factors in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 5084–5089. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro-Oncology 2016, 18, v1–v75. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Rubin, J.B.; Lathia, J.D.; Berens, M.E.; Barnholtz-Sloan, J.S. Females have the survival advantage in glioblastoma. Neuro-Oncology 2018, 20, 576–577. [Google Scholar] [CrossRef]
- Ostrom, W.; Coleman, W.; Huang, J.B.; Rubin, J.D.; Lathia, M.E.; Beren, G.; Speyer, P.; Liao, M.R.; Wrensch, J.E.; Eckel-Passow, J.E.; et al. Sex-specific gene and pathway modeling of inherited glioma risk. Neuro-Oncology 2019, 21, 71–82. [Google Scholar]
- Li, H.Y.; Sun, C.R.; He, M.; Yin, L.C.; Du, H.G.; Zhang, J.M. Correlation between tumor location and clinical properties of glioblastomas in frontal and temporal lobes. World Neurosurg. 2018, 112, e407–e414. [Google Scholar] [CrossRef]
- Wnuk, A.; Przepiorska, K.; Pietrzak, B.A.; Kajta, M. Emerging evidence on membrane estrogen receptors as novel therapeutic targets for central nervous system pathologies. Int. J. Mol. Sci. 2023, 24, 4043. [Google Scholar] [CrossRef] [PubMed]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchhri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen receptor bta (ERbeta): A ligand activated tumor suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ge, N.; Guan, X.; Han, C.; Li, Y.; Shen, L.; Chen, M.; Zhang, B.; Qu, C.; Zou, W. The location of estrogen receptor variant ER-α36 is associated with the invasion of glioblastoma. Steroids 2023, 194, 109224. [Google Scholar] [CrossRef]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the past, present, and future of anti-angiogenic therapy in glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef]
- Lin, H.Y.; Liao, K.H.; Ko, C.Y.; Chen, G.Y.; Hsu, S.P.; Hung, C.Y.; Hsu, T.I. 17β-Estradiol induces temozolomide resistance through NRF2-mediated redox homeostasis in glioblastoma. Free Radic. Biol. Med. 2021, 172, 430–440. [Google Scholar] [CrossRef]
- Honikl, L.S.; Lammer, F.; Gempt, J.; Meyer, B.; Schlegel, J.; Delbridge, C. High expression of estrogen receptor alpha and aromatase in glial tumor cells is associated with gender-independent survival benefits in glioblastoma patients. J. Neuroncol. 2020, 147, 567–575. [Google Scholar] [CrossRef]
- Santellan-Hernandez, J.O.; Alvarez-Castro, J.A.; Aguilar-Hidalgo, K.M.; Soto, F.C.; Escalante, J.R.; Ichikawa-Escamilla, E.; Silva, M.J.A.; Mejia-Perez, S.I. Multifocal glioblastoma and hormone replacement therapy in a transgender female. Surg. Neurol. Int. 2023, 14, 106. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2009. [Google Scholar]
- Tebbi, C.K. Etiology of Acute Leukemia: A Review. Cancers 2021, 13, 2256. [Google Scholar] [CrossRef]
- Leeman, D.S.; Brunet, A. Stem cells: Sex specificity in the blood. Nature 2014, 505, 488–490. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Vargas-Delgado, M.E.; Meier, L.; Loges, S. Sexual dimorphism in solid and hematological malignancies. Semin. Immunopathol. 2019, 41, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Locatelli, S.L.; Pagano, M.T.; Ascione, B.; Careddu, G.; Dupuis, M.L.; Marconi, M.; Carlo-Stella, C.; Malorni, W.; Matarrese, P.; et al. The natural estrogen receptor beta agonist silibinin as a promising therapeutic tool in diffuse large B-cell Lymphoma. Anticancer Res. 2022, 42, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Yakimchuk, K.; Hasni, M.S.; Guan, J.; Chao, M.P.; Sander, B.; Okret, S. Inhibition of lymphoma vascularization and dissemination by estrogen receptor beta agonists. Blood 2014, 123, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lam, S.; Mak, J.C.; Zheng, C.; Ho, J.C. Erlotinib-Induced Autophagy in Epidermal Growth Factor Receptor Mutated Non-SmallCell Lung Cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.K.; Subathra Devi, C. Molecular and therapeutic insights of rapamycin: A multi-faceted drug from Streptomyces hygroscopicus. Mol. Biol. Rep. 2023, 50, 3815–3833. [Google Scholar] [CrossRef]
- Benvenuto, M.; Mattera, R.; Masuelli, L.; Taffera, G.; Andracchio, O.; Tresoldi, I.; Lido, P.; Giganti, M.G.; Godos, J.; Modesti, A.; et al. (±)-Gossypol induces apoptosis and autophagy in head and neck carcinoma cell lines and inhibits the growth of salivary gland tumor cells transplanted into BALB/c mice. Int. J. Food Sci. Nutr. 2017, 68, 298–312. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and negatively impacts osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef]
- Liu, P.F.; Tsai, K.L.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chang, H.W.; Shiau, C.W.; Goan, Y.G.; Tseng, H.H.; Wu, C.H.; et al. Drug repurposing screen identifies tioconazole as an ATG4 inhibitor that suppresses autophagy and sensitizes tumor cells to chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef]
- Bhat, P.; Kriel, J.; Shubha Priya, B.; Basappa; Shivananju, N.S.; Loos, B. Modulation of autophagy in cancer therapy: Advances and challenges in sensitizing tumor cell death. Biochem. Pharmacol. 2018, 147, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ni, Z.; Yan, X.; Dai, X.; Hu, C.; Zheng, Y.; He, F.; Lian, J. Targeting the MIR34C-5p-ATG4B-autophagy axis increases the sensitivity of cervical cancer cells to pirarubicin. Autophagy 2016, 12, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, C.; Matteoni, S.; Matarrese, P.; Signore, M.; Ascione, B.; Iessi, E.; Gurtner, A.; Sacconi, A.; Ricci-Vitiani, L.; Pallini, R.; et al. Chlorpromazine affects glioblastoma bioenergetics by interfering with pyruvate kinase M2. Cell Death Dis. 2023, 14, 821. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, Y.; Jing, L.; Claret, F.X.; Li, Q.; Tian, T.; Liang, X.; Ruan, Z.; Jiang, L.; Yao, Y.; et al. Autophagy in the “inflammation-carcinogenesis” pathway of liver immunotherapy and HCC. Cancer Lett. 2017, 411, 82–89. [Google Scholar] [CrossRef]
- Attwood, K.M.; Robichaud, A.; Westhaver, L.P.; Castle, E.L.; Brandman, D.M.; Balgi, A.D.; Roberge, M.; Colp, P.; Croul, S.; Kim, I.; et al. Raloxifene prevents stress granule dissolution, impairs translational control and promotes cell death during hypoxia in glioblastoma cells. Cell Death Dis. 2020, 11, 989. [Google Scholar] [CrossRef]
- Bailly, C. Molecular and cellular basis of the anticancer activity of the prenylated flavonoid icaritin in hepatocellular carcinoma. Chem. Biol. Interact. 2020, 325, 109124. [Google Scholar] [CrossRef]
- Anobile, D.P.; Salaroglio, I.C.; Tabbò, F.; La Vecchia, S.; Akman, M.; Napoli, F.; Bungaro, M.; Benso, F.; Aldieri, E.; Bironzo, P.; et al. Autocrine 17-β-Estradiol/estrogen receptor-α loop determines the response to immune checkpoint inhibitors in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 3958–3973. [Google Scholar] [CrossRef]
- Barrera-Rodriguez, R.; Morales-Fuentes, J. Lung cancer in women. Lung Cancer 2012, 3, 79–89. [Google Scholar]
- Schafer, J.M.; Xiao, T.; Kwon, H.; Collier, K.; Chang, Y.; Abdel-Hafiz, H.; Bolyard, C.; Chung, D.; Yang, Y.; Sundi, D.; et al. Sex-biased adaptive immune regulation in cancer development and therapy. Science 2022, 25, 104717. [Google Scholar] [CrossRef]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 13, 2163–2170. [Google Scholar] [CrossRef]
- Kammula, A.V.; Schäffer, A.A.; Rajagopal, P.S.; Kurzrock, R.; Ruppin, E. Outcome differences by sex in oncology clinical trials. Nat. Commun. 2024, 15, 2608. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phases Autophagy | ERα | ERβ | ERα/β |
|---|---|---|---|
| Phagophore Induction | ATG2A, PI3KC, ATG9A | UVRAG | Ambra1, ULK2, ATG14, ATG13 |
| Phagophore Expansion | ATG5, LC3B, SQSTM1, ATG4C, ATG4D, ATG7 | ATG10, ATG16L1 | ATG4B, ATG10, ATG16L2, ATG7 |
| Fusion | TECPR1, FYCO1 |
| Colorectal Cancer | Gastric Cancer | Lung Cancer | Glioblastoma | Hematological Malignancies (NHL, HL, L, MM) | |
|---|---|---|---|---|---|
| Incidence absolute numbers | 538,262 | 135,610 | 484,306 | 67,559 | 306,921 |
| Sex disparity in incidence M/F ratio | 1.3 | 1.4 | 2.0 | 1.2 | 1.2 * |
| Mortality absolute numbers | 247,842 | 95,431 | 375,569 | 54,001 | 228,961 |
| Sex disparity in mortality M/F ratio | 1.2 | 1.4 | 2.0 | 1.2 | 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vona, R.; Cittadini, C.; Ortona, E.; Matarrese, P. Sex Disparity in Cancer: Role of Autophagy and Estrogen Receptors. Cells 2025, 14, 273. https://doi.org/10.3390/cells14040273
Vona R, Cittadini C, Ortona E, Matarrese P. Sex Disparity in Cancer: Role of Autophagy and Estrogen Receptors. Cells. 2025; 14(4):273. https://doi.org/10.3390/cells14040273
Chicago/Turabian StyleVona, Rosa, Camilla Cittadini, Elena Ortona, and Paola Matarrese. 2025. "Sex Disparity in Cancer: Role of Autophagy and Estrogen Receptors" Cells 14, no. 4: 273. https://doi.org/10.3390/cells14040273
APA StyleVona, R., Cittadini, C., Ortona, E., & Matarrese, P. (2025). Sex Disparity in Cancer: Role of Autophagy and Estrogen Receptors. Cells, 14(4), 273. https://doi.org/10.3390/cells14040273