
Mitogen-Activated Protein Kinase Phosphatase-2 Deletion Promotes Hyperglycemia and Susceptibility to Streptozotocin-Induced Diabetes in Female Mice In Vivo


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents, Antibodies, and Immunoblotting
2.2. Animal Studies
2.2.1. Animal Study Design 1a
2.2.2. Animal Study Design 1b
2.2.3. Experimental Unit 1c
2.2.4. Sample Size 2
2.2.5. Inclusion and Exclusion Criteria 3
2.2.6. Randomization 4
2.2.7. Blinding 5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Genotype | Gender | N | |
|---|---|---|---|---|
| T2D Treated | Streptozotocin | MKP-2 WT | F | 12 |
| Streptozotocin | MKP-2 KO | F | 12 | |
| Streptozotocin | MKP-2 WT | M | 10 | |
| Streptozotocin | MKP-2 KO | M | 10 | |
| T2D Control | Citrate buffer | MKP-2 WT | F | 11 |
| Citrate buffer | MKP-2 KO | F | 11 | |
| Citrate buffer | MKP-2 WT | M | 10 | |
| Citrate buffer | MKP-2 KO | M | 10 | |
| T1D Treated | Streptozotocin | MKP-2 WT | F | 9 |
| Streptozotocin | MKP-2 KO | F | 8 | |
| Streptozotocin | MKP-2 WT | M | 13 | |
| Streptozotocin | MKP-2 KO | M | 12 | |
| T1D Control | Citrate buffer | MKP-2 WT | F | 9 |
| Citrate buffer | MKP-2 KO | F | 7 | |
| Citrate buffer | MKP-2 WT | M | 11 | |
| Citrate buffer | MKP-2 KO | M | 11 |
2.2.8. Outcome Measures 6
2.2.9. Statistical Methods 7
2.2.10. Experimental Animals 8a
2.2.11. Genotype 8b
2.2.12. Experimental Procedures 9a
| Procedures | Resources |
|---|---|
Pharmacological procedure
| Mouse model |
Blood glucose measurement
| Reagents Primer used for real-time PCR (see Section 2.5) Antibodies used for the immunoblotting:
cDNA Reagent Kit: Thermo Fisher (Waltham, MA, USA) (#4368814) |
Euthanasia
| Equipment and Software Equipment used:
Software used: Prism 9 statistical software (GraphPad Software, La Jolla, CA, USA). |
2.2.13. Experimental Procedures 9b
2.3. Immunoblotting
2.4. RNA Extraction and Real-Time PCR Analysis
2.5. Histological Analysis of Tissue Sections
3. Results
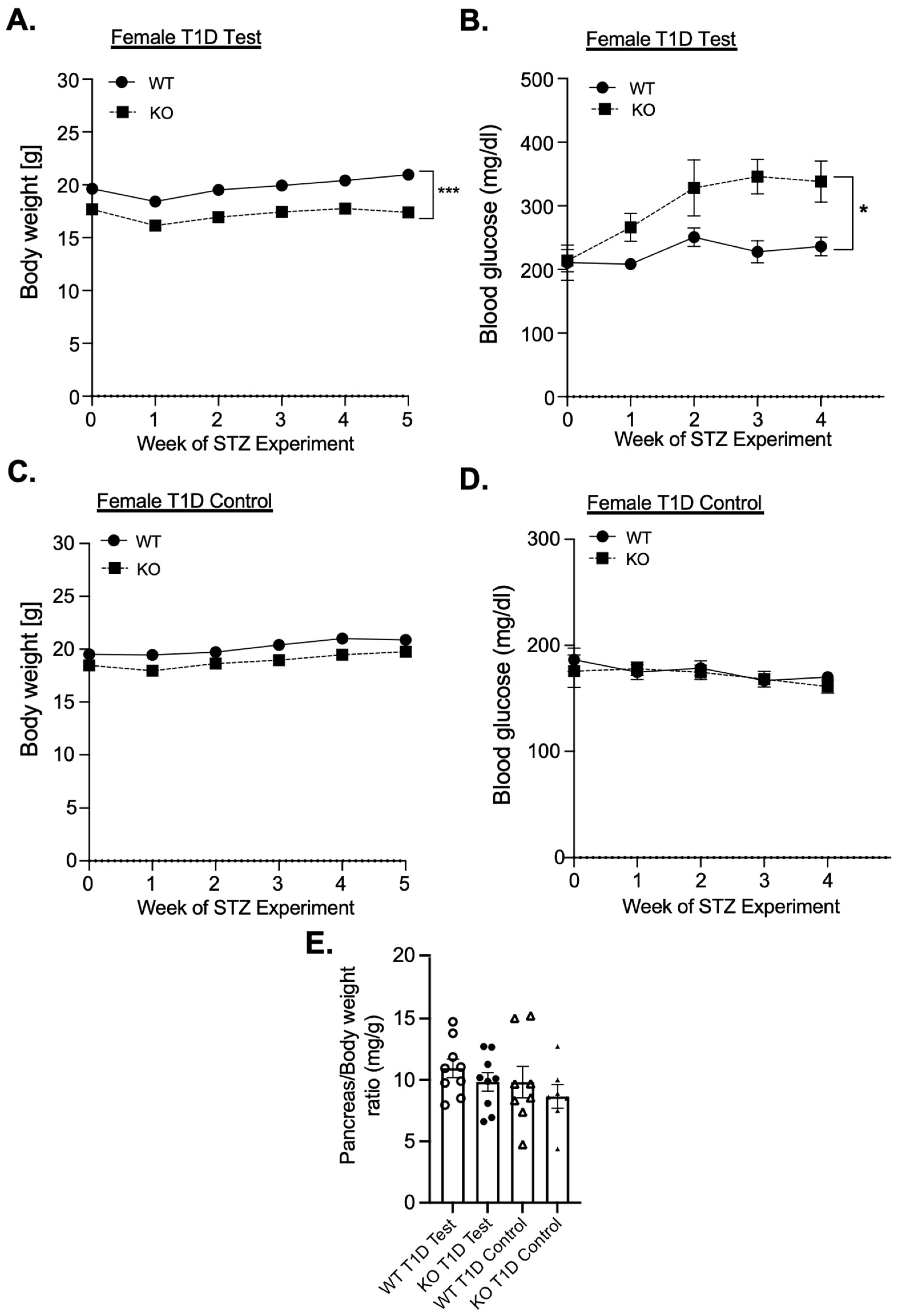
3.1. Female MKP-2 KO Are Hyperglycemic and Susceptible to STZ-Induced Diabetes
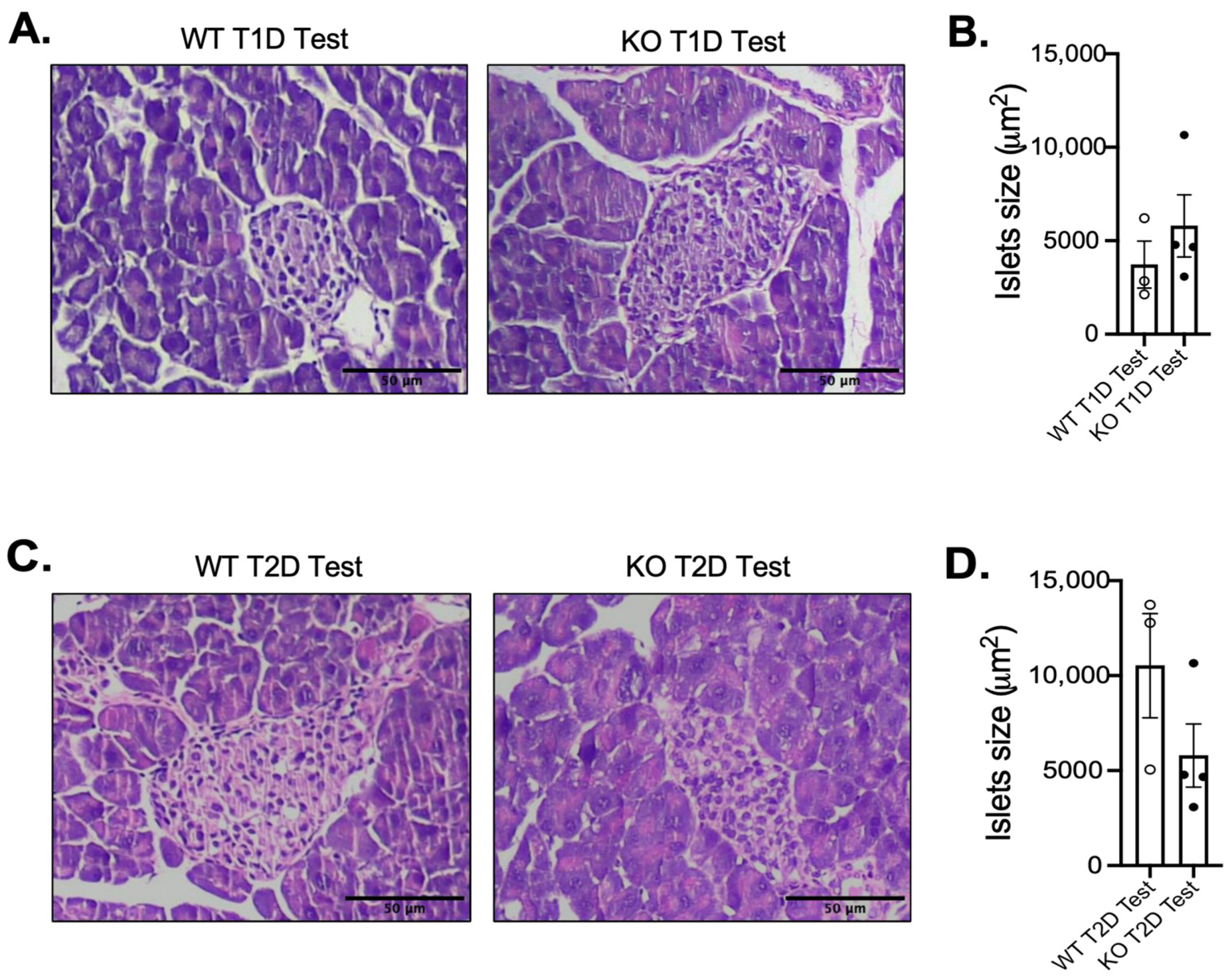
3.2. Impact of MKP-2 Deficiency in Islets of Female MKP-2 KO Mice in Diabetes
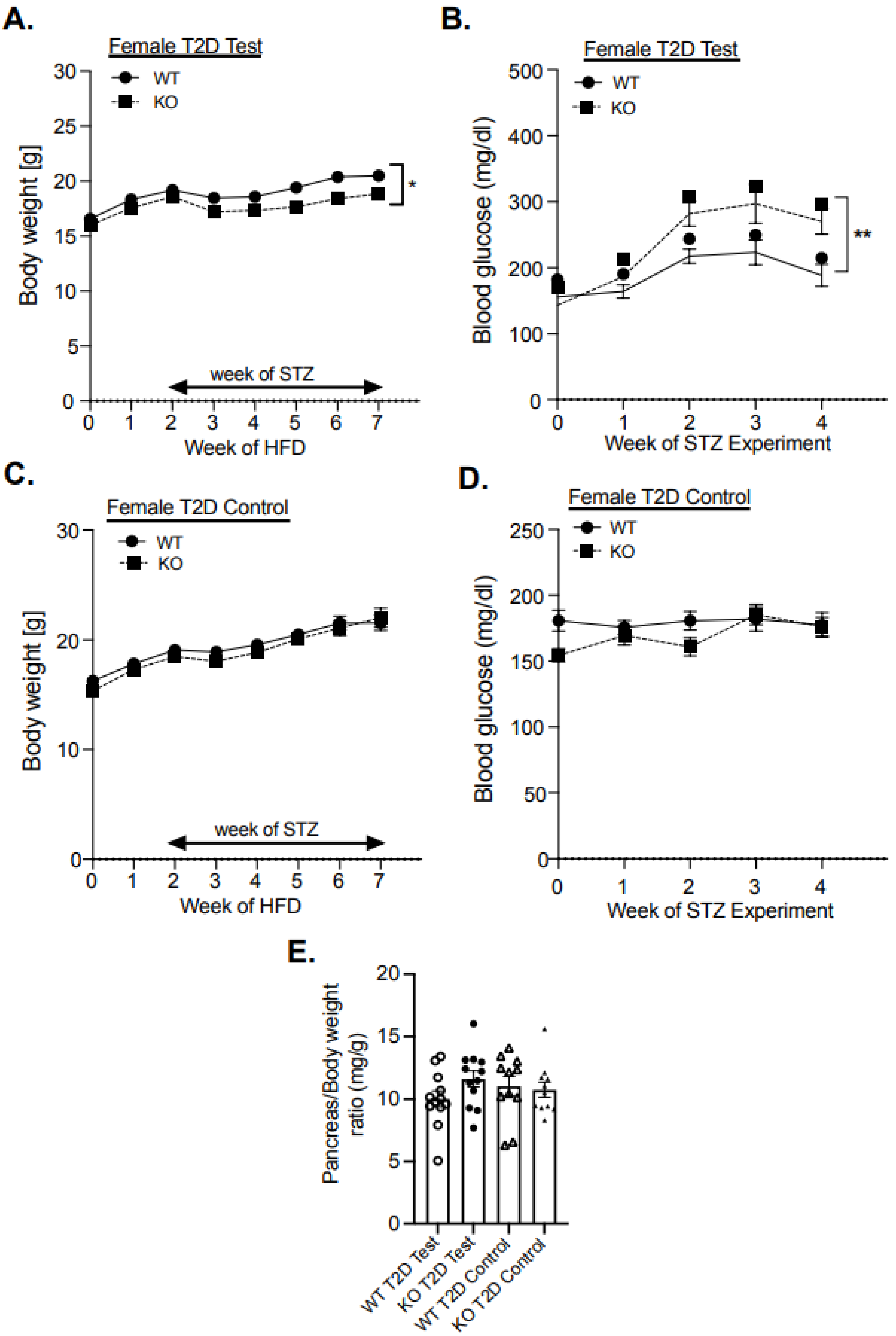
3.3. Enhanced MAPK Activity and Suppression of Pancreatic Development Gene Expression in Female MKP-2 KO Mice in T2D
3.4. Increased Adiposity and Insulin Resistance in Female MKP-2 KO Mice in T2D
4. Discussion
Limitations of This Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glovaci, D.; Fan, W.; Wong, N.D. Epidemiology of Diabetes Mellitus and Cardiovascular Disease. Curr. Cardiol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, T.; Brinks, R.; Isom, S.; Dabelea, D.; Divers, J.; Mayer-Davis, E.J.; Lawrence, J.M.; Pihoker, C.; Dolan, L.; Liese, A.D.; et al. Projections of Type 1 and Type 2 Diabetes Burden in the U.S. Population Aged < 20 Years Through 2060: The SEARCH for Diabetes in Youth Study. Diabetes Care 2023, 46, 313–320. [Google Scholar] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Sanyal, A.J.; Zhang, S.; Hartman, M.L.; Bue-Valleskey, J.M.; Hoogwerf, B.J.; Haupt, A. Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Sicree, R.A.; Zimmet, P.Z.; Dunstan, D.W.; Cameron, A.J.; Welborn, T.A.; Shaw, J.E. Differences in height explain gender differences in the response to the oral glucose tolerance test—The AusDiab study. Diabet. Med. 2008, 25, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes Among Adults in the United States, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Brazzale, A.R.; Moro, E.; Vrbíková, J.; Bendlova, B.; Sbrignadello, S.; Tura, A.; Pacini, G. Influence of increasing BMI on insulin sensitivity and secretion in normotolerant men and women of a wide age span. Obesity 2012, 20, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Marchese, E.; Rodeghier, C.; Monson, R.S.; McCracken, B.; Shi, T.; Schrock, W.; Martellotto, J.; Oberholzer, J.; Danielson, K.K. Enumerating β-Cells in Whole Human Islets: Sex Differences and Associations with Clinical Outcomes After Islet Transplantation. Diabetes Care 2015, 38, e176–e177. [Google Scholar] [CrossRef] [PubMed]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef]
- Daniels Gatward, L.F.; Kennard, M.R.; Smith, L.I.F.; King, A.J.F. The use of mice in diabetes research: The impact of physiological characteristics, choice of model and husbandry practices. Diabet. Med. 2021, 38, e14711. [Google Scholar] [CrossRef] [PubMed]
- Sidarala, V.; Kowluru, A. The Regulatory Roles of Mitogen-Activated Protein Kinase (MAPK) Pathways in Health and Diabetes: Lessons Learned from the Pancreatic β-Cell. Recent. Pat. Endocr. Metab. Immune Drug Discov. 2017, 10, 76–84. [Google Scholar] [CrossRef]
- Šrámek, J.; Němcová-Fürstová, V.; Kovář, J. Kinase Signaling in Apoptosis Induced by Saturated Fatty Acids in Pancreatic β-Cells. Int. J. Mol. Sci. 2016, 17, 1400. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and Obesity-Related Cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Youl, E.; Bardy, G.; Magous, R.; Cros, G.; Sejalon, F.; Virsolvy, A.; Richard, S.; Quignard, J.F.; Gross, R.; Petit, P.; et al. Quercetin potentiates insulin secretion and protects INS-1 pancreatic β-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol. 2010, 161, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef]
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002. [Google Scholar] [CrossRef]
- Seternes, O.M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef]
- Lawan, A.; Shi, H.; Gatzke, F.; Bennett, A.M. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol. Life Sci. 2013, 70, 223–237. [Google Scholar] [CrossRef]
- Wu, Z.; Jiao, P.; Huang, X.; Feng, B.; Feng, Y.; Yang, S.; Hwang, P.; Du, J.; Nie, Y.; Xiao, G.; et al. MAPK phosphatase-3 promotes hepatic gluconeogenesis through dephosphorylation of forkhead box O1 in mice. J. Clin. Invest. 2010, 120, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, B.; Eberlé, D.; Suzuki, R.; Kahn, C.R. Overexpression of the dual-specificity phosphatase MKP-4/DUSP-9 protects against stress-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 3545–3550. [Google Scholar] [CrossRef]
- Zhao, T.; Ma, J.; Li, L.; Teng, W.; Tian, Y.; Ma, Y.; Wang, W.; Yan, W.; Jiao, P. MKP-5 Relieves Lipotoxicity-Induced Islet β-Cell Dysfunction and Apoptosis via Regulation of Autophagy. Int. J. Mol. Sci. 2020, 21, 7161. [Google Scholar] [CrossRef] [PubMed]
- Fernando, S.; Sellers, J.; Smith, S.; Bhogoju, S.; Junkins, S.; Welch, M.; Willoughby, O.; Ghimire, N.; Secunda, C.; Barmanova, M.; et al. Metabolic Impact of MKP-2 Upregulation in Obesity Promotes Insulin Resistance and Fatty Liver Disease. Nutrients 2022, 14, 2475. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, M.S.; Cadalbert, L.C.; McGachy, H.A.; Shweash, M.; Schroeder, J.; Kurnik, M.; Sloss, C.M.; Bryant, C.E.; Alexander, J.; Plevin, R. MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana. PLoS Pathog. 2010, 6, e1001192. [Google Scholar] [CrossRef]
- Lawan, A.; Al-Harthi, S.; Cadalbert, L.; McCluskey, A.G.; Shweash, M.; Grassia, G.; Grant, A.; Boyd, M.; Currie, S.; Plevin, R. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J. Biol. Chem. 2011, 286, 12933–12943. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. BMJ Open Sci. 2020, 4, e100115. [Google Scholar] [PubMed]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef] [PubMed]
- Chao, P.C.; Li, Y.; Chang, C.H.; Shieh, J.P.; Cheng, J.T.; Cheng, K.C. Investigation of insulin resistance in the popularly used four rat models of type-2 diabetes. Biomed. Pharmacother. 2018, 101, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Mason, J.; Beaudet, A.L.; Benjamini, Y.; Bower, L.; Braun, R.E.; Brown, S.D.M.; Chesler, E.J.; Dickinson, M.E.; Flenniken, A.M.; et al. Prevalence of sexual dimorphism in mammalian phenotypic traits. Nat. Commun. 2017, 8, 15475. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Zhang, L.; Gatzke, F.; Min, K.; Jurczak, M.J.; Al-Mutairi, M.; Richter, P.; Camporez, J.P.; Couvillon, A.; Pesta, D.; et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol. Cell Biol. 2015, 35, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.; Gavin, T.P.; Shulman, G.I.; Fernandez-Hernando, C.; et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef]
- Ramzy, M.M.; El-Sheikh, A.A.; Kamel, M.Y.; Abdelwahab, S.A.; Morsy, M.A. Mechanism of testicular protection of carvedilol in streptozotocin-induced diabetic rats. Indian. J. Pharmacol. 2014, 46, 161–165. [Google Scholar] [PubMed]
- Carbone, L.; Carbone, E.T.; Yi, E.M.; Bauer, D.B.; Lindstrom, K.A.; Parker, J.M.; Austin, J.A.; Seo, Y.; Gandhi, A.D.; Wilkerson, J.D. Assessing cervical dislocation as a humane euthanasia method in mice. J. Am. Assoc. Lab. Anim. Sci. 2012, 51, 352–356. [Google Scholar]
- Huang, X.; Jiang, J.; Shen, J.; Xu, Z.; Gu, F.; Pei, J.; Zhang, L.; Tang, P.; Yin, P. The Influences of Cryopreservation Methods on RNA, Protein, Microstructure and Cell Viability of Skeletal Muscle Tissue. Biopreserv Biobank 2024, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Lessieur, E.M.; Du, Y.; Liu, H.; Kern, T.S. Successful induction of diabetes in mice demonstrates no gender difference in development of early diabetic retinopathy. PLoS ONE 2020, 15, e0238727. [Google Scholar] [CrossRef] [PubMed]
- Manaer, T.; Yu, L.; Zhang, Y.; Xiao, X.J.; Nabi, X.H. Anti-diabetic effects of shubat in type 2 diabetic rats induced by combination of high-glucose-fat diet and low-dose streptozotocin. J. Ethnopharmacol. 2015, 169, 269–274. [Google Scholar] [CrossRef]
- Zhao, T.; Tian, Y.; Zhao, J.; Sun, D.; Ma, Y.; Wang, W.; Yan, W.; Jiao, P.; Ma, J. Loss of mitogen-activated protein kinase phosphate-5 aggravates islet dysfunction in mice with type 1 and type 2 diabetes. FASEB J. 2024, 38, e23437. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The MAFB transcription factor impacts islet α-cell function in rodents and represents a unique signature of primate islet β-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E91–E102. [Google Scholar] [CrossRef]
- Lee, Y.S.; Morinaga, H.; Kim, J.J.; Lagakos, W.; Taylor, S.; Keshwani, M.; Perkins, G.; Dong, H.; Kayali, A.G.; Sweet, I.R.; et al. The fractalkine/CX3CR1 system regulates β cell function and insulin secretion. Cell 2013, 153, 413–425. [Google Scholar] [CrossRef]
- Cheon, H.; Cho, J.M.; Kim, S.; Baek, S.H.; Lee, M.K.; Kim, K.W.; Yu, S.W.; Solinas, G.; Kim, S.S.; Lee, M.S. Role of JNK activation in pancreatic beta-cell death by streptozotocin. Mol. Cell Endocrinol. 2010, 321, 131–137. [Google Scholar] [CrossRef]
- Von Bank, H.; Kirsh, C.; Simcox, J. Aging adipose: Depot location dictates age-associated expansion and dysfunction. Ageing Res. Rev. 2021, 67, 101259. [Google Scholar] [CrossRef] [PubMed]
- Soltani, N.; Qiu, H.; Aleksic, M.; Glinka, Y.; Zhao, F.; Liu, R.; Li, Y.; Zhang, N.; Chakrabarti, R.; Ng, T.; et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. USA 2011, 108, 11692–11697. [Google Scholar] [CrossRef]
- Li, Z.; Shangguan, Z.; Liu, Y.; Wang, J.; Li, X.; Yang, S.; Liu, S. Puerarin protects pancreatic β-cell survival via PI3K/Akt signaling pathway. J. Mol. Endocrinol. 2014, 53, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, A.; Unek, G.; Kipmen-Korgun, D.; Korgun, E.T. The PI3K/Akt and MAPK-ERK1/2 pathways are altered in STZ induced diabetic rat placentas. Histol. Histopathol. 2014, 29, 743–756. [Google Scholar] [PubMed]
- Hulmi, J.J.; Silvennoinen, M.; Lehti, M.; Kivelä, R.; Kainulainen, H. Altered REDD1, myostatin, and Akt/mTOR/FoxO/MAPK signaling in streptozotocin-induced diabetic muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E307–E315. [Google Scholar] [CrossRef]
- Goedeke, L.; Perry, R.J.; Shulman, G.I. Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Labarbuta, R.; Tortosa, F.; Biondi, G.; Marrano, N.; Peschechera, A.; Carchia, E.; Orlando, M.R.; Leonardini, A.; Cignarelli, A.; et al. Exendin-4 protects pancreatic beta cells from palmitate-induced apoptosis by interfering with GPR40 and the MKK4/7 stress kinase signalling pathway. Diabetologia 2013, 56, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, L.E.; Lawrence, J.M.; Isom, S.; Jensen, E.T.; Dabelea, D.; Liese, A.D.; Dolan, L.M.; Shah, A.S.; Bellatorre, A.; Sauder, K.; et al. Trends in incidence of youth-onset type 1 and type 2 diabetes in the USA, 2002–2018: Results from the population-based SEARCH for Diabetes in Youth study. Lancet Diabetes Endocrinol. 2023, 11, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Venditti, M.; Romano, M.Z.; Boccella, S.; Haddadi, A.; Biasi, A.; Maione, S.; Minucci, S. Type 1 diabetes impairs the activity of rat testicular somatic and germ cells through NRF2/NLRP3 pathway-mediated oxidative stress. Front. Endocrinol. 2024, 15, 1399256. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Casado, L.; Juncos-Tobarra, M.A.; Cháfer-Rudilla, M.; de Onzoño, L.; Blázquez-Cabrera, J.A.; Miralles-García, J.M. Effect of experimental diabetes and STZ on male fertility capacity. Study in rats. J. Androl. 2010, 31, 584–592. [Google Scholar] [CrossRef]
- Xu, Y.; Lei, H.; Guan, R.; Gao, Z.; Li, H.; Wang, L.; Song, W.; Gao, B.; Xin, Z. Studies on the mechanism of testicular dysfunction in the early stage of a streptozotocin induced diabetic rat model. Biochem. Biophys. Res. Commun. 2014, 450, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Gourdy, P.; Guillaume, M.; Fontaine, C.; Adlanmerini, M.; Montagner, A.; Laurell, H.; Lenfant, F.; Arnal, J.F. Estrogen receptor subcellular localization and cardiometabolism. Mol. Metab. 2018, 15, 56–69. [Google Scholar] [CrossRef]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Lawan, A.; Bennett, A.M. Mitogen-Activated Protein Kinase Regulation in Hepatic Metabolism. Trends Endocrinol. Metab. 2017, 28, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.H.; Ramesh, S.; Liao, P.H.; Kuo, W.W.; Chen, M.C.; Kuo, C.H.; Li, C.C.; Wang, T.F.; Lin, Y.M.; Lin, Y.J.; et al. Phosphorylation of Bcl-2 by JNK confers gemcitabine resistance in lung cancer cells by reducing autophagy-mediated cell death. Environ. Toxicol. 2023, 38, 2121–2131. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Carstensen, M.; Ouwens, D.M. Anti-inflammatory cytokines and risk of type 2 diabetes. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 39–50. [Google Scholar] [CrossRef] [PubMed]
- Elias, I.; Franckhauser, S.; Ferré, T.; Vilà, L.; Tafuro, S.; Muñoz, S.; Roca, C.; Ramos, D.; Pujol, A.; Riu, E.; et al. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes 2012, 61, 1801–1813. [Google Scholar] [CrossRef]
- Gannon, M.; Kulkarni, R.N.; Tse, H.M.; Mauvais-Jarvis, F. Sex differences underlying pancreatic islet biology and its dysfunction. Mol. Metab. 2018, 15, 82–91. [Google Scholar] [CrossRef]
- Handgraaf, S.; Dusaulcy, R.; Visentin, F.; Philippe, J.; Gosmain, Y. 17-β Estradiol regulates proglucagon-derived peptide secretion in mouse and human α- and L cells. JCI Insight 2018, 3, e98569. [Google Scholar] [CrossRef]
- Rettberg, J.R.; Yao, J.; Brinton, R.D. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 2014, 35, 8–30. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghimire, N.; Welch, M.; Secunda, C.; Fink, A.; Lawan, A. Mitogen-Activated Protein Kinase Phosphatase-2 Deletion Promotes Hyperglycemia and Susceptibility to Streptozotocin-Induced Diabetes in Female Mice In Vivo. Cells 2025, 14, 261. https://doi.org/10.3390/cells14040261
Ghimire N, Welch M, Secunda C, Fink A, Lawan A. Mitogen-Activated Protein Kinase Phosphatase-2 Deletion Promotes Hyperglycemia and Susceptibility to Streptozotocin-Induced Diabetes in Female Mice In Vivo. Cells. 2025; 14(4):261. https://doi.org/10.3390/cells14040261
Chicago/Turabian StyleGhimire, Nabin, Morgan Welch, Cassandra Secunda, Alexis Fink, and Ahmed Lawan. 2025. "Mitogen-Activated Protein Kinase Phosphatase-2 Deletion Promotes Hyperglycemia and Susceptibility to Streptozotocin-Induced Diabetes in Female Mice In Vivo" Cells 14, no. 4: 261. https://doi.org/10.3390/cells14040261
APA StyleGhimire, N., Welch, M., Secunda, C., Fink, A., & Lawan, A. (2025). Mitogen-Activated Protein Kinase Phosphatase-2 Deletion Promotes Hyperglycemia and Susceptibility to Streptozotocin-Induced Diabetes in Female Mice In Vivo. Cells, 14(4), 261. https://doi.org/10.3390/cells14040261