
Eugenol Promotes Apoptosis in Leukemia Cells via Targeting the Mitochondrial Biogenesis PPRC1 Gene
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Cell Culture, and Reagents
2.2. Blood Samples
2.3. Cytotoxicity Assay
2.4. Apoptosis Analysis by Annexin V/Flow Cytometry
2.5. RNA Purification and qRT-PCR
| PPRC1: | Forward: 5’-TCTCCCCATCCGAAACACAA-3’ |
| Reverse: 5’-GTCTCTTGGGTCTCAGGCTT-3’ | |
| c-MYC: | Forward: 5’-CCTGGTGCTCCATGAGGAGAC-3’ |
| Reverse: 5’-CAGACTCTCCAGCATCCACT-3’ | |
| GAPDH: | Forward: 5’-GAG TCC ACT GGC GTC TTG-3’ |
| Reverse: 5’-GGG GTG CTA AGC AGT TGG T-3’ |
2.6. Cellular Lysate Preparation and Immunoblotting
2.7. Quantification of Protein Expression Level
2.8. Cell Proliferation
2.9. Mitochondrial Staining
2.10. Data Mining
2.11. Mutation Pathogenicity and Data Visualization
2.12. Statistical Analysis
3. Results
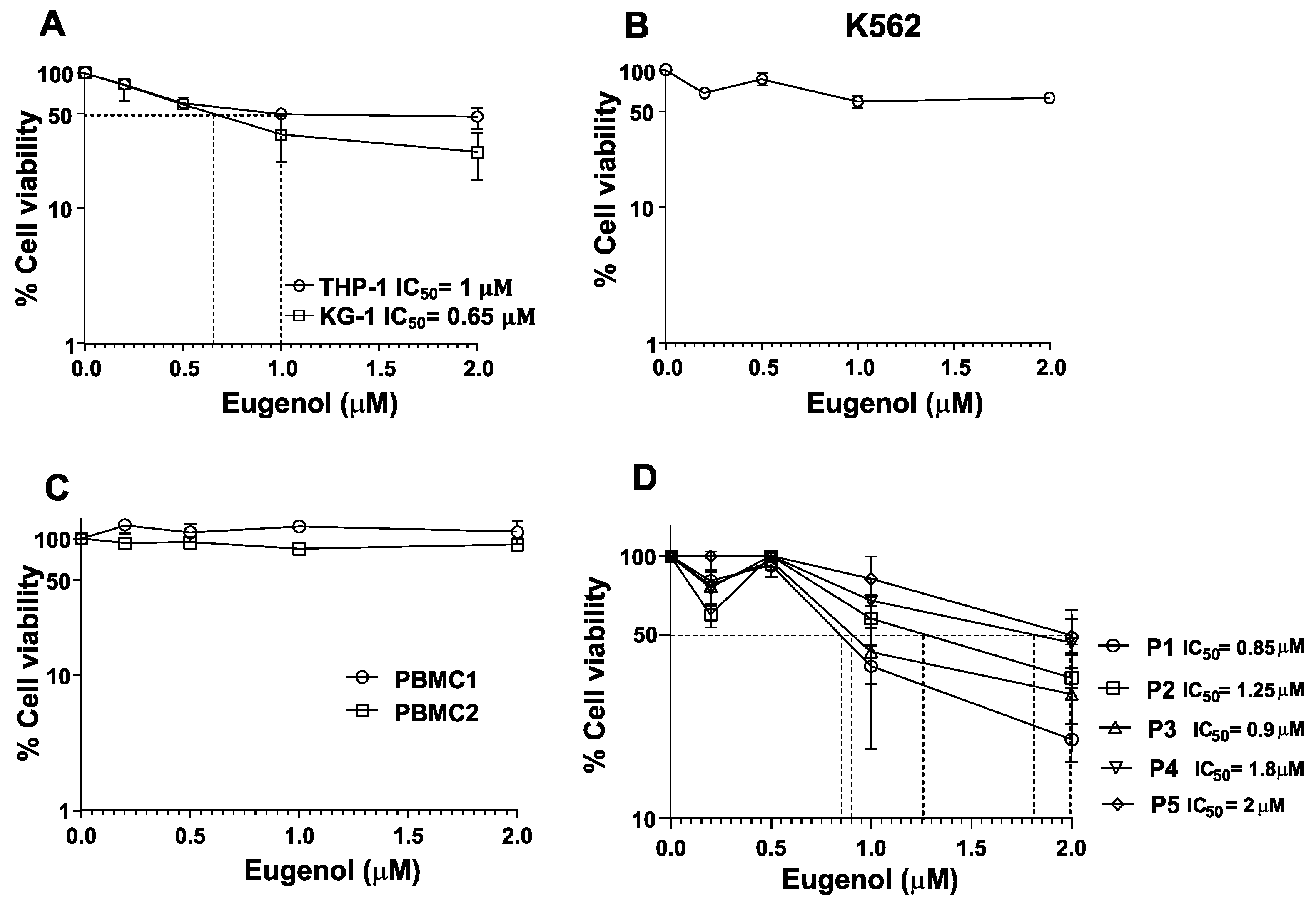
3.1. Leukemia Cell Lines and Primary Cells Are Sensitive to Eugenol
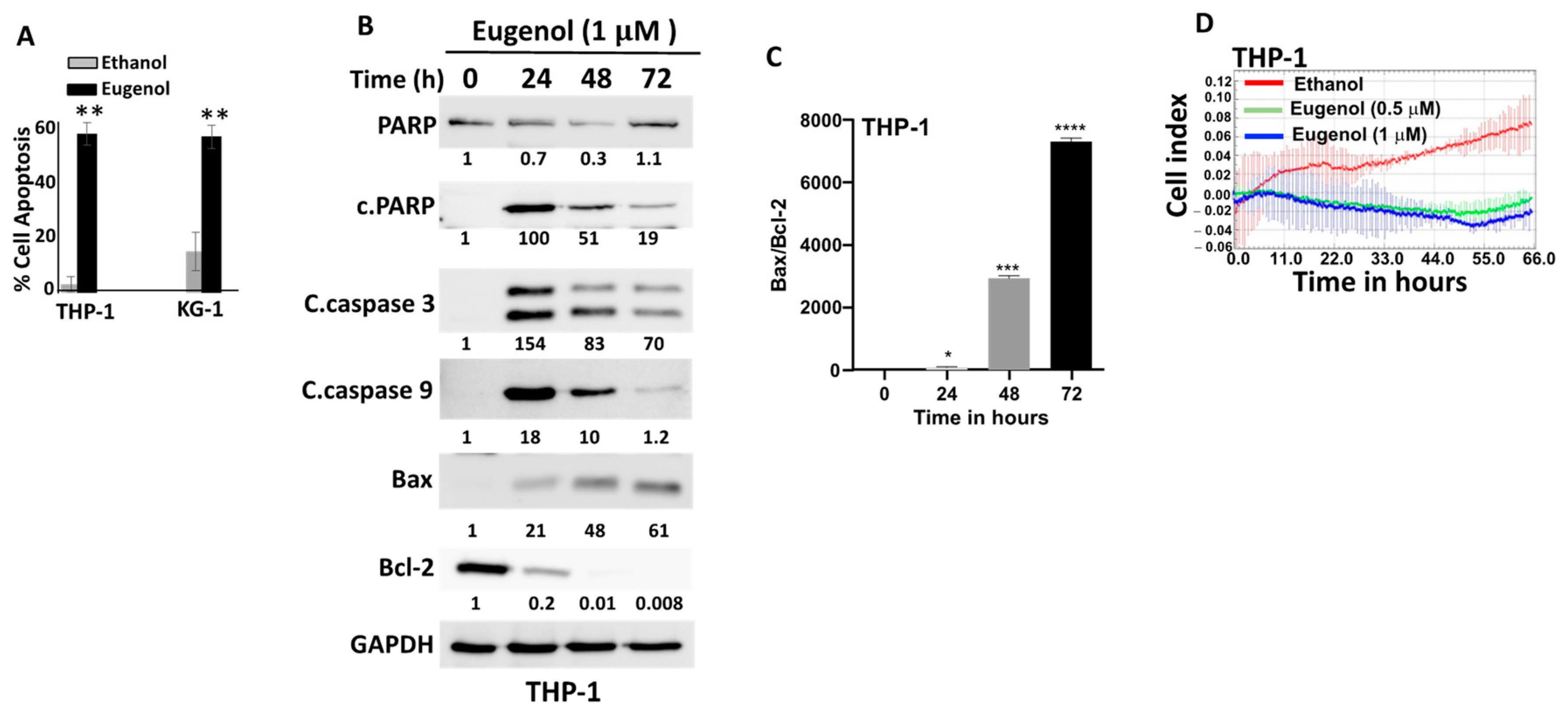
3.2. Eugenol Promotes Apoptosis and Inhibits Proliferation in AML Cells
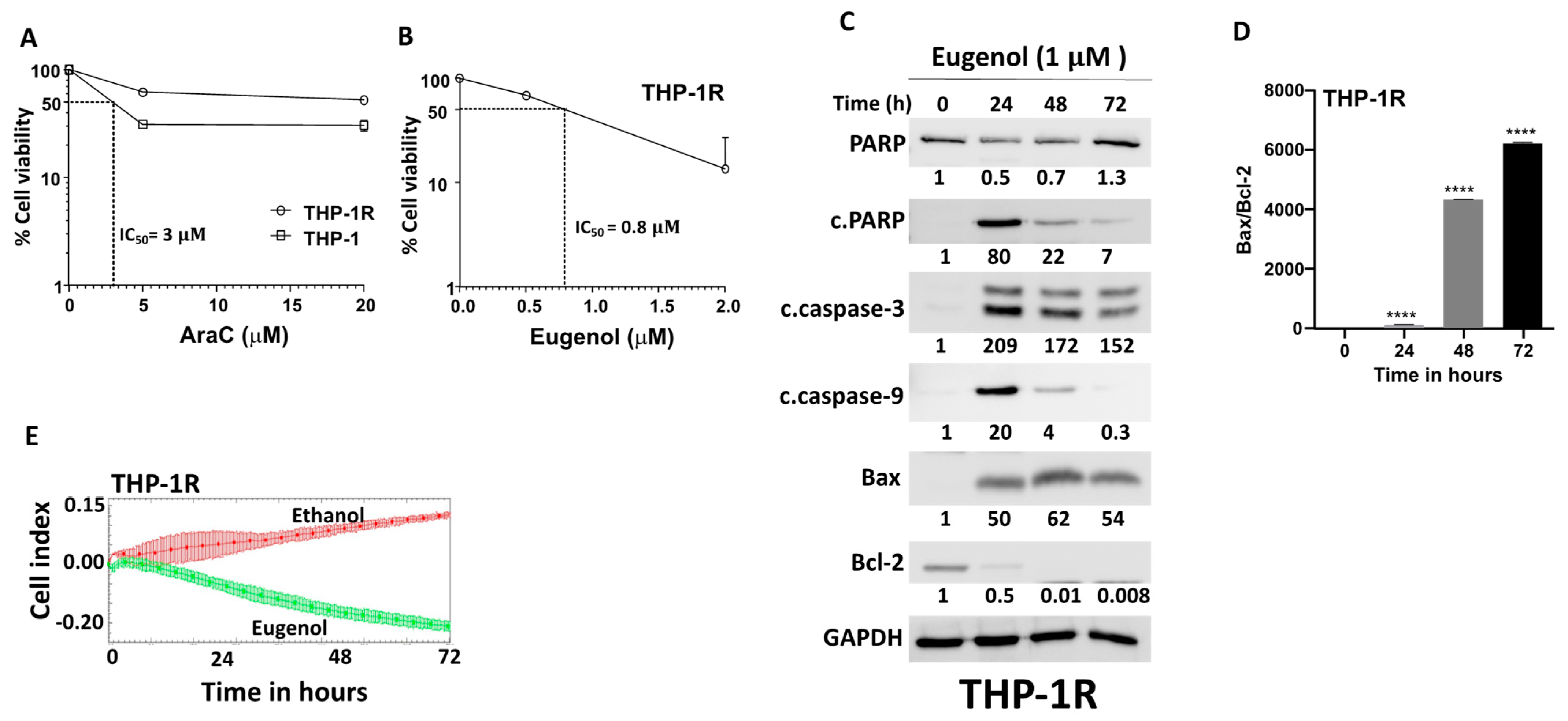
3.3. Eugenol Promotes Apoptosis and Inhibits Proliferation in Cytarabine-Resistant AML Cells
3.4. Eugenol Downregulates c-MYC and PPRC1 in AML Cell Lines
3.5. Eugenol Reduces Mitochondrial Membrane Potential (MMP)
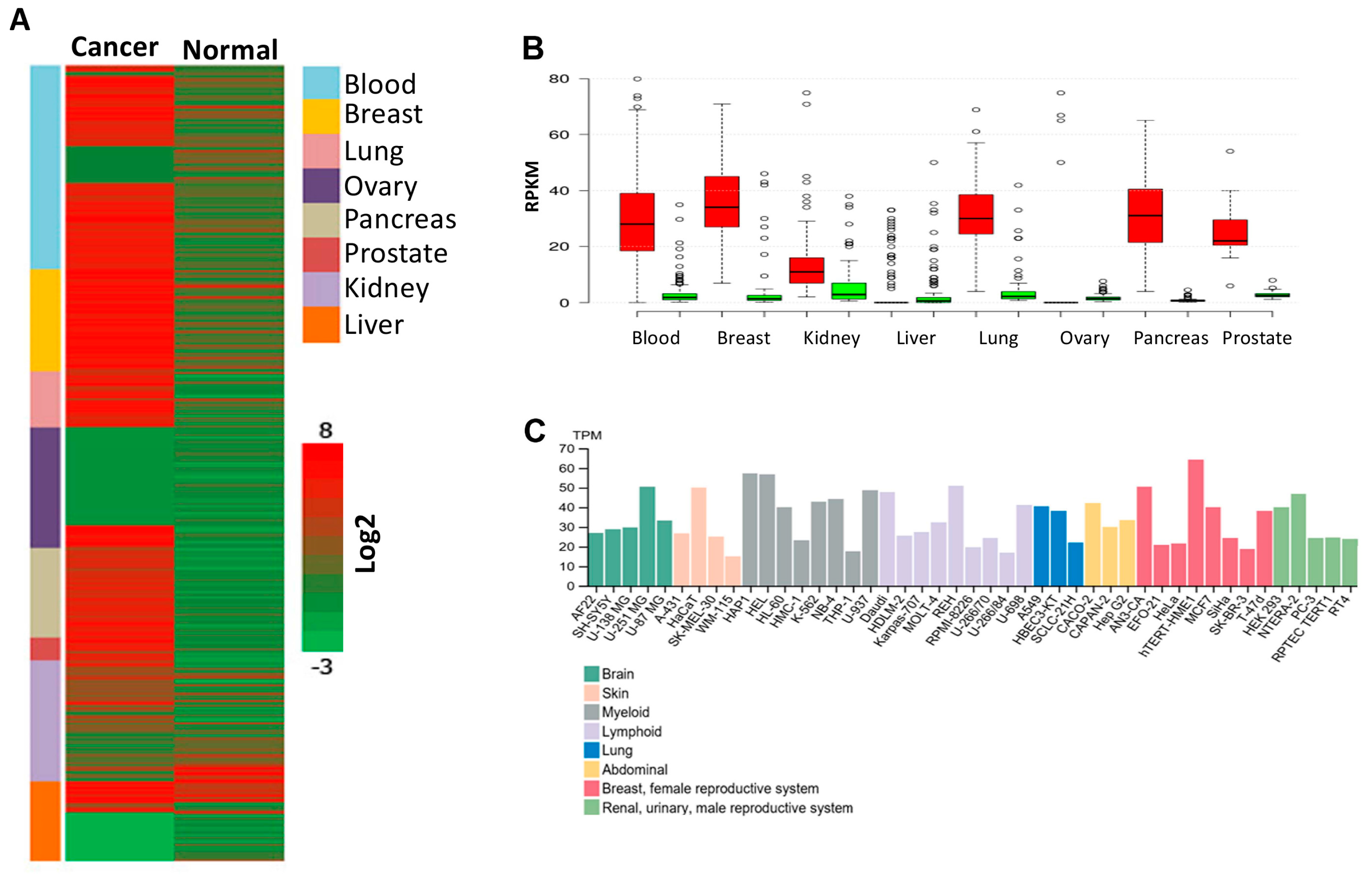
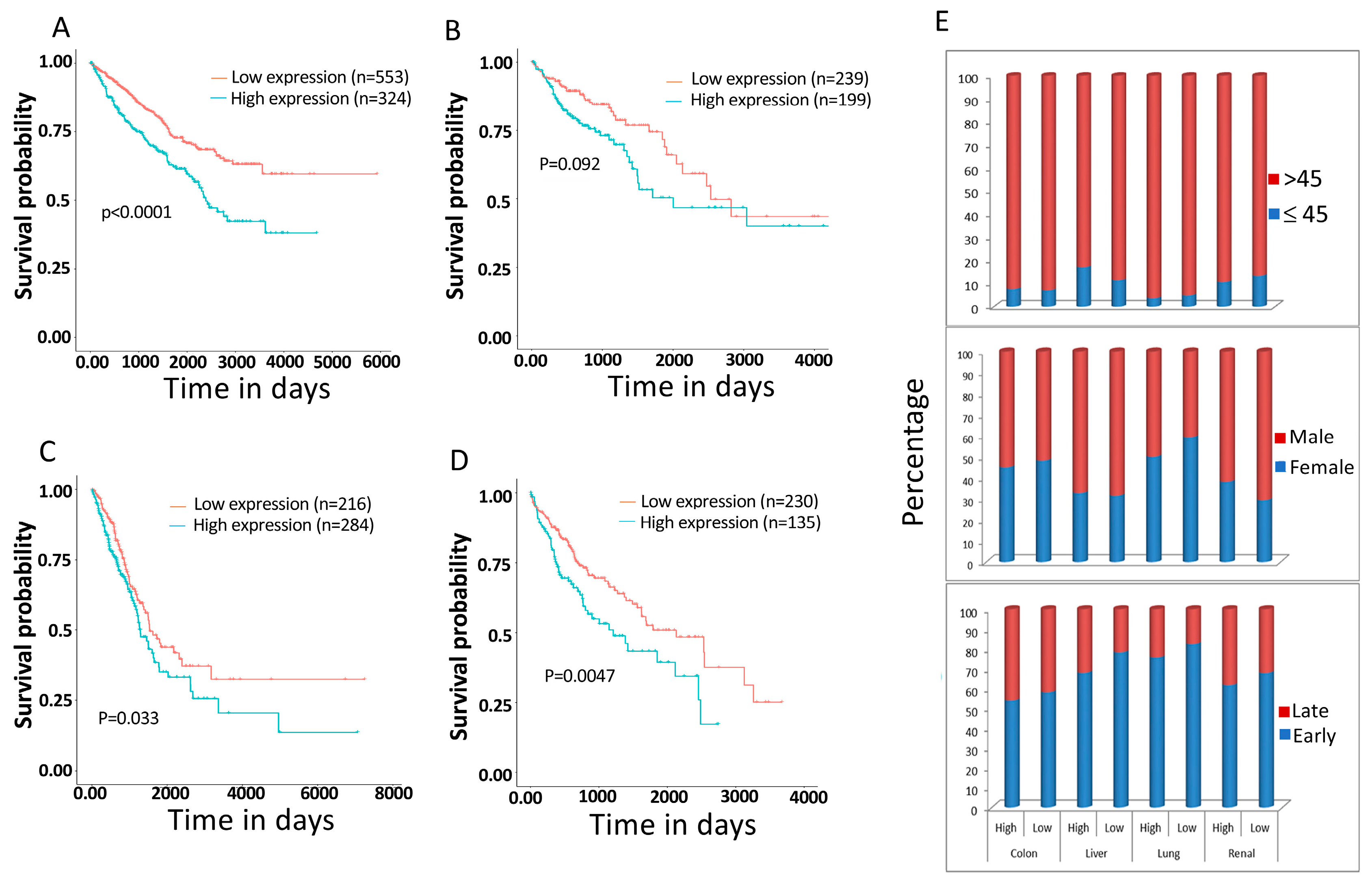
3.6. PPRC1 Expression Is Higher in Cancer Cells Relative to Normal Cells and Significantly Correlates with Short Overall Survival in Several Cancers
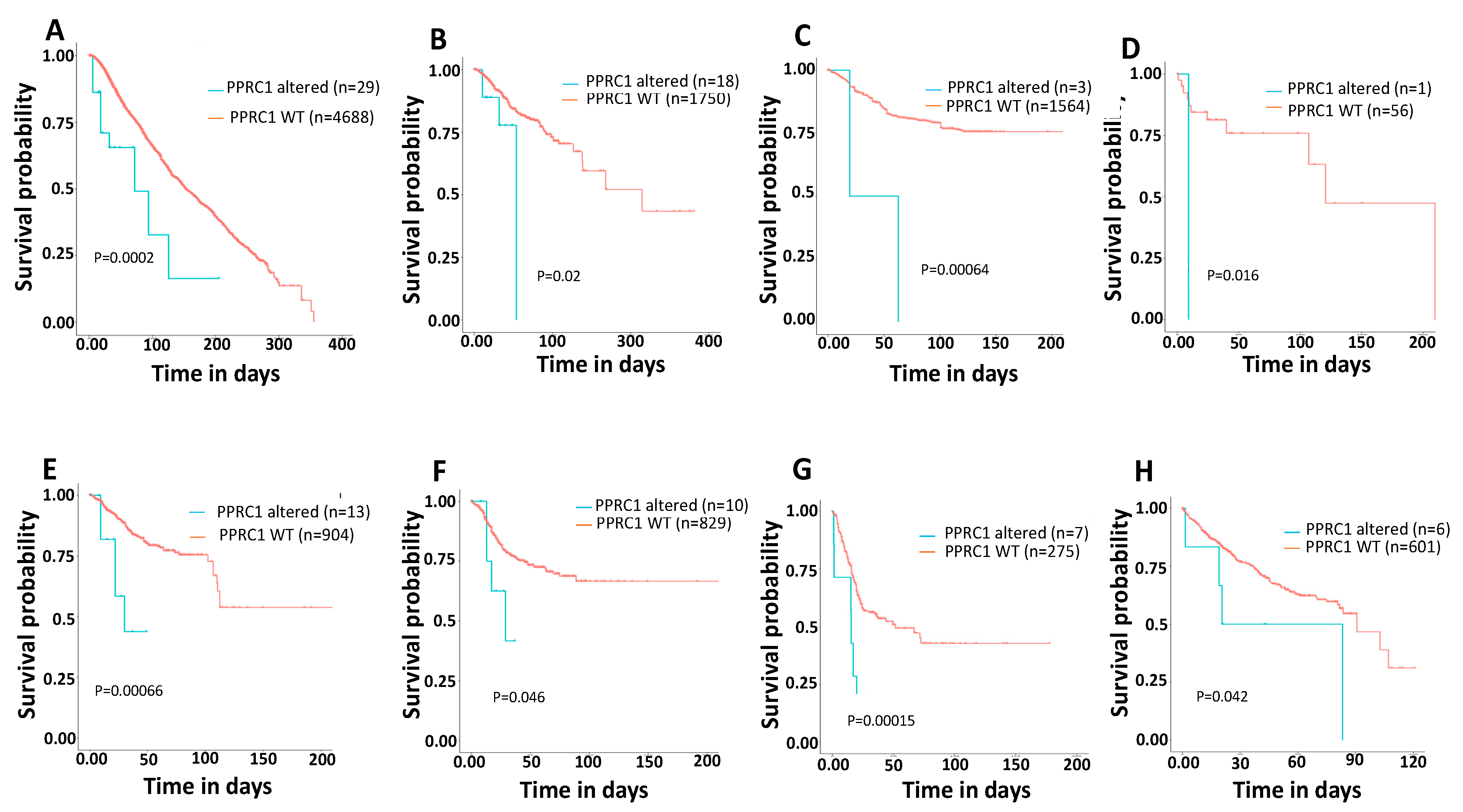
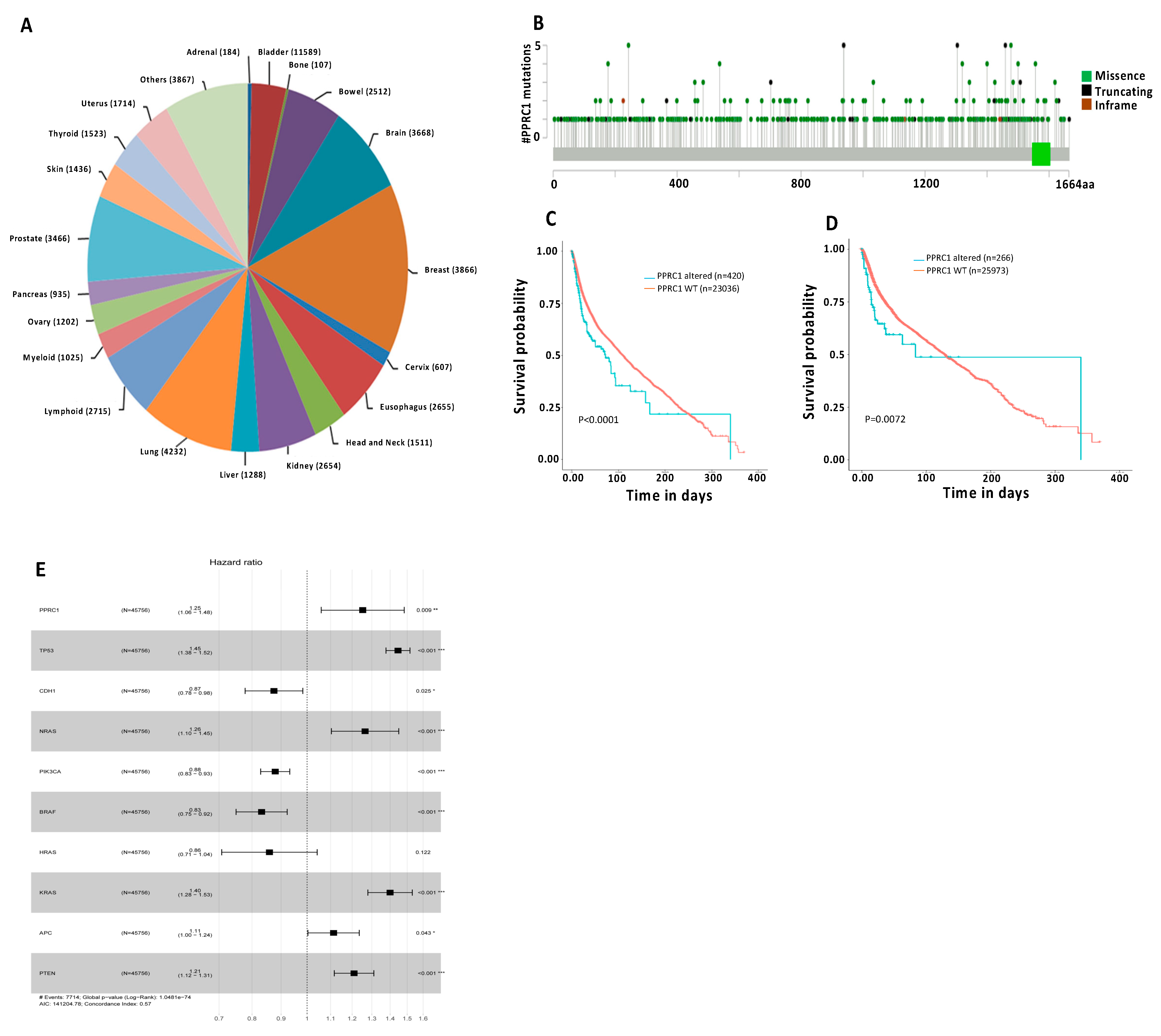
3.7. Genetic Alterations in the PPRC1 Gene Correlate Significantly with Severe Clinical Outcome in Cancer Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MMP | Mitochondrial membrane potential |
| PBMCs | Peripheral Blood Mononuclear cells |
| AML | Acute myeloid leukemia |
| DFS | Disease-free survival |
| OS | Overall survival |
References
- Aberger, F.; Hutterer, E.; Sternberg, C.; Del Burgo, P.J.; Hartmann, T.N. Acute myeloid leukemia—Strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun. Signal 2017, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; St-Pierre, J.; Giguere, V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Andersson, U.; Scarpulla, R.C. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Gleyzer, N.; Scarpulla, R.C. Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J. Biol. Chem. 2009, 284, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- CAncer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J. Cell Sci. 2001, 114 Pt 20, 3591–3598. [Google Scholar] [CrossRef]
- Zari, A.T.; Zari, T.A.; Hakeem, K.R. Anticancer Properties of Eugenol: A Review. Molecules 2021, 26, 7407. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharif, I.; Remmal, A.; Aboussekhra, A. Eugenol triggers apoptosis in breast cancer cells through E2F1/survivin down-regulation. BMC Cancer 2013, 13, 600. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.S.; Al-Sharif, I.; Sultan, A.; Al-Mazrou, A.; Remmal, A.; Aboussekhra, A. Eugenol potentiates cisplatin anti-cancer activity through inhibition of ALDH-positive breast cancer stem cells and the NF-kappaB signaling pathway. Mol. Carcinog. 2018, 57, 333–346. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Vo, T.T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; Deangelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Roberts, A.W. BCL2 Inhibition: A New Paradigm for the Treatment of AML and Beyond. Hemasphere 2023, 7, e912. [Google Scholar] [CrossRef]
- Purhonen, J.; Klefstrom, J.; Kallijarvi, J. MYC-an emerging player in mitochondrial diseases. Front. Cell Dev. Biol. 2023, 11, 1257651. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Hoffman, B.; Amanullah, A.; Shafarenko, M.; Liebermann, D.A. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene 2002, 21, 3414–3421. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Scarpulla, R.C. Concerted Action of PGC-1-related Coactivator (PRC) and c-MYC in the Stress Response to Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 25529–25541. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Savagner, F.; Mirebeau, D.; Jacques, C.; Guyetant, S.; Morgan, C.; Franc, B.; Reynier, P.; Malthiery, Y. PGC-1-related coactivator and targets are upregulated in thyroid oncocytoma. Biochem. Biophys. Res. Commun. 2003, 310, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Mirebeau-Prunier, D.; Le Pennec, S.; Jacques, C.; Gueguen, N.; Poirier, J.; Malthiery, Y.; Savagner, F. Estrogen-related receptor alpha and PGC-1-related coactivator constitute a novel complex mediating the biogenesis of functional mitochondria. FEBS J. 2010, 277, 713–725. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Name | Mutation Type | Cancer Type |
|---|---|---|
| P941Tfs | Frameshift insertion | Stomach Adenocarcinoma |
| P941Tfs | Frameshift insertion | Renal Clear Cell Carcinoma |
| P941Tfs | Frameshift insertion | Prostate Adenocarcinoma |
| P941Tfs | Frameshift insertion | Breast Invasive Ductal Carcinoma |
| P941Tfs | Frameshift insertion | Colorectal Adenocarcinoma |
| P941Tfs | Frameshift insertion | Small Cell Lung Cancer |
| P941Tfs | Frameshift insertion | Glioblastoma |
| P941fs | Frameshift deletion | HEC151 cell line, Endometrial adenocarcinoma |
| P941fs | Frameshift deletion | HEC6 cell line, Endometrial endometrioid adenocarcinoma |
| P941fs | Frameshift deletion | CCK81 cell line, Colon adenocarcinoma |
| P941fs | Frameshift deletion | SNU-1 cell line, Gastric carcinoma |
| P938fs | Frameshift insertion | 22RV1 cell line, Prostate carcinoma |
| P938fs | Frameshift insertion | HEC-265 cell line, Endometrial adenocarcinoma |
| P938fs | Frameshift insertion | MDAPCA2B cell line, Prostate carcinoma |
| P938fs | Frameshift insertion | SNU407 cell line, Colon adenocarcinoma |
| P938fs | Frameshift insertion | CW-2 cell line, Colon adenocarcinoma |
| P938fs | Frameshift insertion | EFO27 cell line, Ovarian mucinous adenocarcinoma |
| R243H/C | Missense | Prostate Adenocarcinoma |
| Lung Squamous Cell Carcinoma | ||
| Uveal Melanoma | ||
| Uterine Endometrioid Carcinoma | ||
| Colon Adenocarcinoma | ||
| P940Hfs | Frameshift deletion | Prostate Adenocarcinoma |
| Stomach Adenocarcinoma | ||
| Uterine Endometrioid Carcinoma | ||
| Colorectal Adenocarcinoma | ||
| R177C | Missense | Uterine Endometrioid Carcinoma |
| Colon Adenocarcinoma | ||
| Bladder Urothelial Carcinoma | ||
| Stomach Adenocarcinoma | ||
| P537S/L/T | Missense | Cutaneous Squamous Cell Carcinoma |
| Cutaneous Melanoma | ||
| Melanoma | ||
| Hepatocellular Adenoma | ||
| R1320*/P/Q | Nonsense/Missense | Lung Adenocarcinoma |
| Cutaneous Melanoma | ||
| Bladder Urothelial Carcinoma | ||
| Cutaneous Squamous Cell Carcinoma | ||
| R1400Q/L/W | Missense | Adenoid Cystic Carcinoma |
| Colorectal Adenocarcinoma | ||
| Serous Ovarian Cancer | ||
| Cutaneous Melanoma | ||
| R1400P | KCL22 cell line, Chronic myelogenous leukemia | |
| R1556Q/* | Missense/Nonsense | Pancreatic Adenocarcinoma |
| Uterine Endometrioid Carcinoma | ||
| Colorectal Adenocarcinoma | ||
| Cutaneous Melanoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Harbi, S.; Alkholiwy, E.M.A.; Ali Ahmed, S.O.; Aljurf, M.; Al-Hejailan, R.; Aboussekhra, A. Eugenol Promotes Apoptosis in Leukemia Cells via Targeting the Mitochondrial Biogenesis PPRC1 Gene. Cells 2025, 14, 260. https://doi.org/10.3390/cells14040260
Al-Harbi S, Alkholiwy EMA, Ali Ahmed SO, Aljurf M, Al-Hejailan R, Aboussekhra A. Eugenol Promotes Apoptosis in Leukemia Cells via Targeting the Mitochondrial Biogenesis PPRC1 Gene. Cells. 2025; 14(4):260. https://doi.org/10.3390/cells14040260
Chicago/Turabian StyleAl-Harbi, Sayer, Elham M. A. Alkholiwy, Syed Osman Ali Ahmed, Mahmoud Aljurf, Reem Al-Hejailan, and Abdelilah Aboussekhra. 2025. "Eugenol Promotes Apoptosis in Leukemia Cells via Targeting the Mitochondrial Biogenesis PPRC1 Gene" Cells 14, no. 4: 260. https://doi.org/10.3390/cells14040260
APA StyleAl-Harbi, S., Alkholiwy, E. M. A., Ali Ahmed, S. O., Aljurf, M., Al-Hejailan, R., & Aboussekhra, A. (2025). Eugenol Promotes Apoptosis in Leukemia Cells via Targeting the Mitochondrial Biogenesis PPRC1 Gene. Cells, 14(4), 260. https://doi.org/10.3390/cells14040260