Highlights
What are the main findings?
- Medium-chain fatty acid decanoic acid (C10) is efficiently metabolized in both wild-type and 5xFAD mouse brain slices, particularly in astrocytes, supporting mitochondrial function.
- Astrocytic acetate metabolism is impaired in 5xFAD mice and familial AD astrocytes exhibit genotype-dependent metabolic responses to C10, with partial alterations in oxidative glucose metabolism in APP and PSEN1 variants.
What are the implications of the main findings?
- Altered astrocytic metabolism might occur before glucose hypometabolism in the 5xFAD mouse model.
- C10 supplementation may provide an auxiliary fuel source capable of supporting brain energy metabolism in Alzheimer’s disease, in part by promoting oxidative metabolism in astrocytes.
Abstract
Alzheimer’s disease (AD) is increasingly recognized as a disorder of cerebral energy metabolism, where impaired glucose utilization contributes to disease pathology. Medium-chain fatty acids (MCFAs), such as decanoic acid (C10), have emerged as promising metabolic substrates due to their ability to bypass glycolytic deficits and support mitochondrial function. In this study, we investigated the metabolic impact of C10 in the 5xFAD mouse model of AD and in human induced pluripotent stem cell (hiPSC)-derived astrocytes carrying familial AD mutations. Utilizing stable 13C-labeled metabolic tracers, we demonstrated that while [U-13C]glucose metabolism was largely preserved in cortical slices of 6-month-old 5xFAD female mice, [1,2-13C]acetate uptake was significantly reduced, suggesting impaired astrocytic metabolism. [U-13C]C10 was efficiently metabolized in both WT and 5xFAD brain slices, particularly in astrocytes, as indicated by high labeling of glutamine and citrate. Furthermore, C10 competitively inhibited glucose and acetate metabolism, suggesting its potential as an auxiliary energy substrate. In hiPSC-derived astrocytes, AD-specific metabolic responses to C10 varied by mutation, with only partial alterations in oxidative glucose metabolism observed in APP and PSEN1 variants, highlighting genotype-dependent metabolic alterations. While AD-related mutations in the hiPSC models did not lead to robust deficits, the in vivo environment in the 5xFAD model is associated with measurable metabolic changes in astrocytes. These findings underscore astrocytic metabolic dysfunction in AD and suggest that C10 supplementation may restore brain energy by supporting astrocytic oxidative metabolism.
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a loss of memory, cognitive decline, and behavioral changes, which is largely driven by metabolic dysfunction, including impaired cerebral glucose metabolism [1,2]. Although the pathogenesis of AD involves multiple factors, including the accumulation of amyloid-beta plaques, neurofibrillary tangles, and neuroinflammation, emerging research increasingly positions AD as a metabolic disorder of the brain, where energy failure significantly contributes to disease progression [3,4,5]. This view has supported a focus on metabolic interventions to tackle AD pathology, especially the use of alternative energy substrates such as medium-chain fatty acids (MCFAs), including decanoic acid (C10) and octanoic acid (C8) [6].
MCFAs are unique due to their ability to bypass complex digestion pathways and directly enter the liver and brain, where they may act as signaling molecules or undergo oxidation to produce ketone bodies. These MCFAs and ketone bodies, including β-hydroxybutyrate, may serve as alternative energy sources, especially when brain glucose metabolism is compromised, a common trait in AD [7]. Many studies have shown that diets enriched with medium-chain triglycerides (MCTs) containing C8 and C10 can improve cognitive function and brain energy metabolism in individuals with mild cognitive impairment (MCI) and AD [8,9,10].
The metabolism of MCFAs in the brain primarily occurs in astrocytes [11], which play a crucial role in supporting neuronal function. Metabolism of C8 and C10 in astrocytes supports glutamine synthesis, which is then used by neurons to synthesize the neurotransmitter GABA [12,13]. This metabolic coupling between astrocytes and neurons is essential for maintaining amino acid and neurotransmitter balance as well as overall brain health [14]. Additionally, MCFAs have been shown to reduce oxidative stress and inflammation, further contributing to their neuroprotective effects [15,16,17].
Among the MCFAs, C10 has gained attention for its distinct metabolic and potential neuroprotective effects [18,19]. C10 influences not only mitochondrial β-oxidation but also cytosolic pathways, stimulating fatty acid synthesis and potentially enhancing neuronal metabolism. The role of C10 in supporting astrocyte metabolism and fatty acid synthesis may also provide neuroprotective effects. Additionally, we recently showed that C10 rescues differences in AMPA-mediated calcium rises in hippocampal astrocytes and neurons in the 5xFAD mouse model of AD pathology [20].
Despite these promising findings, the exact mechanisms by which MCFAs, particularly C10, exert their beneficial effects in AD remain to be fully elucidated. Here, we investigated brain energy metabolism in the 5xFAD mouse model using uniformly 13C-labeled glucose ([U-13C]glucose), acetate ([1,2-13C]acetate), and C10 ([U-13C]C10) to probe neuronal and astrocytic metabolic activity in cerebral slices. We also measured the effect of C10 on [U-13C]glucose metabolism in human induced pluripotent stem cell (hiPSC)-derived astrocytes. We demonstrated that astrocytic metabolism was primarily impaired in the cerebral cortex of the 5xFAD model and that C10 could act as an auxiliary brain energy substrate, replacing glucose to some extent. The hiPSC-derived AD astrocytes displayed distinct metabolic alterations in oxidative glucose metabolism upon C10 supplementation; however, these responses varied across AD genotypes, underscoring the complexity of astrocyte metabolic profiles in AD.
2. Materials and Methods
2.1. Materials
The stable 13C isotopes [1,2-13C]acetate (CLM-440-SP, sodium salt, 99%), [U-13C]decanoic acid ([U-13C]C10, CLM-9950-PK, 98%), and [U-13C]glucose (CLM-1396-5, 99%) were all obtained from Cambridge Isotope Laboratories (Tewksbury, MA, USA). Acetate (S5636, sodium salt 99%), decanoic acid (12C10, C1875), and D-glucose (16301) were purchased from Sigma Aldrich (St. Louis, MI, USA). All other chemicals used were of the purest grade available from commercial sources.
2.2. Animals
Female transgenic 5xFAD mice (TG(APPSwFlLon,PSEN1*M146L*L286V)6799Vas, Jax strain: 034840) and wild-type (WT) mice (Jax strain: 100012) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). A colony was bred and maintained at the Department of Drug Design and Pharmacology, University of Copenhagen. The 5xFAD mice express five familial AD mutations in the amyloid precursor protein (APP) and presenillin1 (PSEN1) genes under the neuron-specific Thy1 promoter, leading to rapid brain amyloid deposition [21]. Genotyping of transgenic and WT mice was performed on ear clippings using a standard PCR-based method (JAX protocol 23370), as previously described [13]. For brain slice experiments, female transgenic 5xFAD and WT mice were used at 6 months old, which correlates with increased β-amyloid 42 deposition, cortical and hippocampal plaques, gliosis, and cognitive decline [22]. A total of 12 mice were used in this study, comprising 6 WT and 6 transgenic 5xFAD. The mice were group-housed in individually ventilated cages. All the mice were bred and maintained in a specific pathogen-free, temperature and humidity-controlled environment with a 12 h light/dark cycle. They were given free access to water and chow. The experiments using animals have been reported in compliance with the ARRIVE guidelines.
2.3. Brain Slice Incubations
Acute brain slice incubations were performed following established procedures [23]. Mice were euthanized by cervical dislocation and decapitation, after which the brain was rapidly removed and placed in ice-cold, oxygenated artificial cerebrospinal fluid (ACSF; in mM: 128 NaCl, 25 NaHCO3, 10 D-glucose, 3 KCl, 2 CaCl2, 1.2 MgSO4, and 0.4 KH2PO4; pH 7.4). Cerebral cortices and hippocampi were dissected and sectioned into 350-μm slices using a McIlwain tissue chopper (The Vibratome Company, St. Louis, MI, USA) [13]. Slices were separated under a microscope, and for each incubation condition, two cortical and five hippocampal slices were transferred to custom-made chambers containing 10 mL ACSF at 37 °C, continuously gassed with 95% O2/5% CO2. Slices were allowed to recover for 60 min [23] before exposure to ACSF supplemented with stable 13C-labeled compounds—5 mM [1,2-13C]acetate, 5 mM [U-13C]glucose, or 200 μM [U-13C]C10 (acetate and C10 were supplemented with 5 mM unlabeled D-glucose)—and were then incubated for an additional 60 min. For competition experiments, labelled substrates were co-incubated with their corresponding unlabeled competitors (i.e., [1,2-13C]acetate in the presence of unlabeled 12C10, [U-13C]glucose in the presence of 12C-acetate, and [U-13C]C10 in the presence of 12C-acetate or 12C-glucose for 60 min. Incubations were terminated by transferring slices into ice-cold 70% ethanol, followed by sonication and centrifugation (4000× g, 20 min). Supernatants were collected, lyophilized, and prepared for gas chromatography–mass spectrometry (GC–MS) analysis.
2.4. Cell Lines
AD astrocytes were derived from healthy hiPSCs, in which APP or PSEN1 mutations were introduced via CRISPR-Cas9; all lines were previously characterized [24,25]. The APP Swedish hiPSC line carries a heterozygous KM670/671NL double mutation (BIONi010-C-38, Cell line repository hPSCreg®), the APP London line harbors a heterozygous V717I mutation (BIONi010-C-37, Cell line repository hPSCreg®), and the PSEN1 line contains a heterozygous E280A mutation (BIONi010-C-30, Cell line repository hPSCreg®). The parental hiPSC line BIONi010-C (also known as K3P53, Cell line repository hPSCreg®), in which all above mentioned mutations were introduced, was used as the wild-type (WT) control. All cell lines carry the APOE3/4 genotype. hiPSC lines were plated on Geltrex (Thermo Fisher Scientific, Waltham, MA, USA) and cultured in Essential 8 Flex medium (Thermo Fisher Scientific, Waltham, MA, USA). Medium was changed every 2 days until the cells reached 70–90% confluence.
2.5. Neural Progenitor Cells Generation and Cell Differentiation
Confluent iPSCs were lifted from Geltrex-coated plates using collagenase type IV (Thermo Fisher Scientific, Waltham, MA, USA) and grown in a 3D culture [26,27]. Neural induction and differentiation were achieved through dual inhibition of SMAD signaling using LDN193189 (Stemgent, Beltsville, MD, USA) and SB431542 (Tocris Bioscience, Bristol, UK) to inhibit the BMP and TGFβ pathway. After 7 days of neuronal induction, embryoid bodies (EBs) were plated on Geltrex-coated plates and supplemented with human epidermal growth factor (EGF) (ProSpec, Ness-Ziona, Israel) and murine fibroblast-basic (FGF) (Peprotech, Cranbury, NJ, USA). Neural rosettes were picked at day 10 for passaging and neural progenitor (NPC) expansion.
2.6. Astrocytic Differentiation
The astrocytes were differentiated according to a modified protocol by Shaltouki et al. 2013 [28], as previously described [26]. From day 16 of neural expansion, astrocyte differentiation media (ADM) was added to 90–100% confluent NPCs. ADM contains FGF, human insulin growth factor IGF-1 (IGF) (Peprotech, Cranbury, NJ, USA), Human Activin A recombinant protein (Thermo Fisher Scientific, Waltham, MA, USA), and human Heregulin β-1 (Peprotech, Cranbury, NJ, USA) to promote astrocyte progenitor production. The media was changed every second day until day 25, when gliogenesis was expected to occur, and after which an astrocyte maturation media (AMM) was added. AMM contains growth factors (IGF, Activin A, and Heregulin) and was supplemented with non-essential amino acids, L-Glutamine, sodium pyruvate, and fetal bovine serum (FBS), all purchased from Thermo Fisher Scientific, and L-Ascorbic acid (Sigma-Aldrich, A8960, St. Louis, MI, USA). The astrocytes were matured for 4 weeks prior to conducting metabolic assays. During the maturation period, the astrocytes were passaged biweekly using Accutase. Immunocytochemical characterization of hiPSC-derived astrocytes (Supplementary Methods and Figure S1) was performed as previously described [29].
2.7. HiPSC Derived Astrocyte Incubations
hiPSC-derived astrocytes were plated at 30,000 cells/cm2 in 6-well plates 48 h before conducting metabolic assays. Cells were rinsed with pre-warmed (37 °C) phosphate-buffered saline (PBS) and incubated for 90 min at 37 °C and 5% CO2 in DMEM supplemented with 13C-labeled substrates. Astrocytes received either 2.5 mM [U-13C]glucose alone or [U-13C]glucose together with 200 µM unlabeled C10, corresponding to reported cerebral concentrations following MCT-enriched diets in mice [30,31]. After incubation, the medium was collected, and metabolism was stopped by washing cells with ice-cold PBS. Cells were then lysed and extracted in 70% ethanol, followed by centrifugation at 4 °C (4000× g, 20 min). The resulting supernatants containing soluble metabolites were lyophilized and stored at −20 °C until GC–MS analysis.
2.8. Metabolic Mapping Using Gas Chromatography Coupled to Mass Spectrometry (GC–MS)
Metabolic 13C-enrichment in TCA cycle intermediates was determined GC–MS following established procedures [32]. Lyophilized extracts from brain slices or hiPSC-derived astrocytes were reconstituted in water, acidified, and subjected to two sequential ethanol extractions. Metabolites were further purified using 96% ethanol followed by benzene to obtain the organic phase. The dried extracts were then derivatized using N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (Sigma Aldrich, St. Louis, MI, USA) prior to GC–MS analysis. The samples were analyzed by GC (Agilent Technologies 7820A chromatograph, J&W GC column HP-5MS, parts no. 19091S-433, Santa Clara, CA, USA) linked to MS (Agilent Technologies 5977E mass spectrometer, Santa Clara, CA, USA). The 13C-enrichment was corrected for the natural abundance of 13C by analyzing standards of the unlabeled metabolites of interest. Data are expressed as the percentage of labeling in isotopologues of the form M + X, where M represents the molecular mass of the unlabeled molecule, and X denotes the number of 13C atoms incorporated. In this study, we focused on M + 3 and M + 2 isotopologues, reflecting metabolites generated during direct metabolism or a first turn of the TCA cycle.
2.9. Statistical Analysis
Data are presented as means ± standard deviation (SD), with individual data points shown. A total of 5–6 mice (n = 5–6) were used for the brain slice incubations. Each individual data point represents a biological replicate (obtained from an individual animal). 5–6 technical replicates (n = 5–6) from 3 independent differentiations were used for the hiPSC-derived astrocyte incubation experiments. Statistical analyses were performed using Graphpad v.10.2.2. An outlier removal (ROUT) test was applied to identify any significant outliers. Outliers were removed from the dataset according to a 1% Q-value. Data were first tested for normality using the Shapiro–Wilk test (α = 0.05). Unpaired data were compared using either a two-tailed Welch’s t-test or Mann–Whitney test. The significance level was set at p < 0.05 and is indicated as *, p < 0.05, **, p < 0.01, ***, p < 0.001 ****, p < 0.0001. For details on the statistical analyses, see Supplementary Table S1–S15. Although brain slices from WT and 5xFAD mice were exposed to the same 13C-labeled substrates, randomization was applied to minimize potential bias in sample handling and processing. Mice were randomly selected for each experimental run by the experimenters. Similarly, for hiPSC-derived astrocyte experiments, wells were randomly assigned to treatment conditions. The study did not apply stratification or blocking, as all animals and cell cultures were treated under uniform conditions using standardized protocols. Cage location was not controlled, but uniform housing and standardized protocols were maintained to reduce confounding effects. Blinding was implemented at multiple stages of the study. Investigators conducting the GC–MS analysis and statistical evaluation were blinded to the group allocations. Sample tubes were coded, ensuring that the identity of the treatment groups remained concealed until after data processing was complete. Blinding was not applied during animal allocation or experimental procedures due to logistical constraints.
3. Results
3.1. Acetate Metabolism Is Impaired in Cerebral Cortical Slices of 5xFAD Mice While Glucose Metabolism Is Maintained
To validate changes in cellular energy metabolism in 5xFAD mice compared to WT, acutely isolated cerebral cortical (Figure 1) and hippocampal slices (Figure S2) were incubated in the presence of 13C-labeled energy substrates to functionally explore metabolism. Glucose is the main energy substrate of the brain, and the metabolism of [U-13C]glucose provides an overview of metabolic function in the brain slices [23]. In both the WT and 5xFAD brain slices, [U-13C]glucose was incorporated via glycolysis, giving rise to labeling in lactate (M + 3) and alanine (M + 3) as well as labelled pyruvate, which enters the TCA cycle as acetyl co-enzyme A (M + 2). No difference was observed between 6-month-old WT and 5xFAD mice in the metabolism of [U-13C]glucose in cortical brain slices (Figure 1A). However, reductions in the 13C labeling of alanine, malate, and glutamate were observed in 5xFAD hippocampal (Figure S2) brain slices. When the slices were incubated with [1,2-13C]acetate, a substrate primarily metabolized in astrocytes [33], a marked decrease in 13C enrichment of all TCA cycle intermediates (citrate, succinate, and malate) and derived amino acids (glutamate, glutamine, GABA, and aspartate) was seen in 5xFAD cerebral cortical slices compared to WT (Figure 1B). The largest reduction in 13C-enrichment (%) was observed in malate (p = 0.0064) and the derived amino acids aspartate (p = 0.0075) and glutamine (p = 0.0031). No alterations in hippocampal [1,2-13C]acetate metabolism were detected between WT and 5xFAD mice (Figure S2). These findings indicate a region-specific hampering of acetate metabolism in the cortical astrocytes of 5xFAD mice and glucose metabolism in their hippocampi.
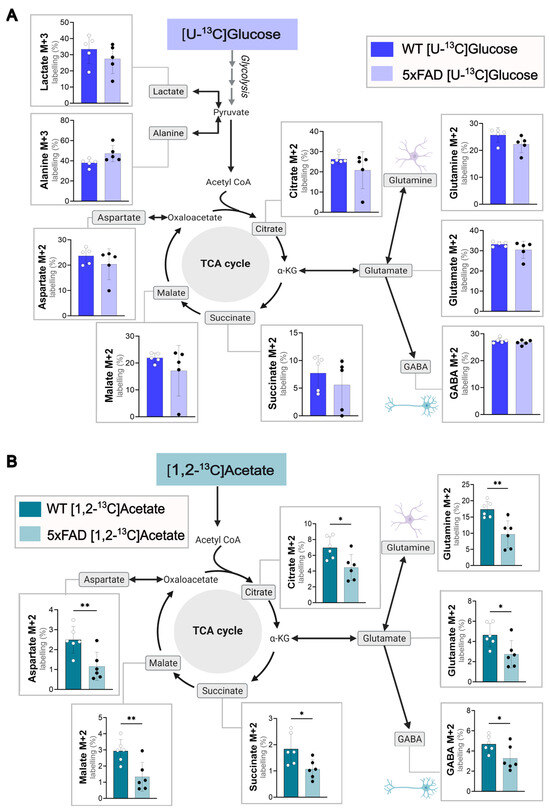
Figure 1.
Glucose and acetate metabolism in WT and 5xFAD mouse cerebral cortex slices. Metabolism of (A) [U-13C]glucose or (B) [1,2-13C]acetate in acutely isolated cerebral cortical slices of 6-month-old WT and 5xFAD mice. Labeling in glycolytic products (M + 3) and first turn (M + 2) TCA cycle intermediates shown. Slices were incubated with medium containing 5 mM [U-13C]glucose or 5 mM [1,2-13C]acetate (supplemented with 5 mM D-glucose), and cell extracts were analyzed via GC-MS. Mean ± SD, n = 5–6 from individual animals, Welch’s t-test or Mann–Whitney test, * p < 0.05, ** p < 0.01.
3.2. Decanoic Acid Can Be Used as a Brain Energy Substrate and Can Compete with Glucose and Acetate to Support Brain Energy Metabolism
C10 can act as an auxiliary brain energy substrate, as demonstrated in previous experiments [12]. Yet the cellular energy metabolism of C10 in the 5xFAD mouse model is still mostly unexplored. To study the differences in C10 metabolism in a pathological setting, acutely isolated cerebral cortical (Figure 2A) and hippocampal slices (Figure S3) from 5xFAD and WT mice were incubated with labeled [U-13C]C10. [U-13C]C10 enters cellular metabolism as 13C2-acetyl CoA units, resulting in 13C-enrichment in TCA cycle intermediates and derived amino acids. Incorporation of 13C due to C10 metabolism was seen in all TCA cycle intermediates and derived amino acids for both wild-type and 5xFAD mice (Figure 2A), verifying the ability of C10 to be used as an energy substrate for metabolism in both mouse genotypes. Interestingly, the highest first turn labeling (%) following metabolism of [U-13C]C10 was seen in the amino acid glutamine M + 2 (WT and 5xFAD; 23.53 ± 1.02% and 23.48 ± 2.00%). This indicates that C10 is metabolized to a higher degree in astrocytes than neurons, as the enzyme GS, which converts glutamate to glutamine, is selectively expressed in astrocytes [34]. High 13C-enrichment (%) was also seen in the TCA cycle intermediate citrate M + 2, which is also suggested to be an indicator for astrocyte metabolism [23,35]. These results support the view that C10 is mainly metabolized in astrocytes. [12]
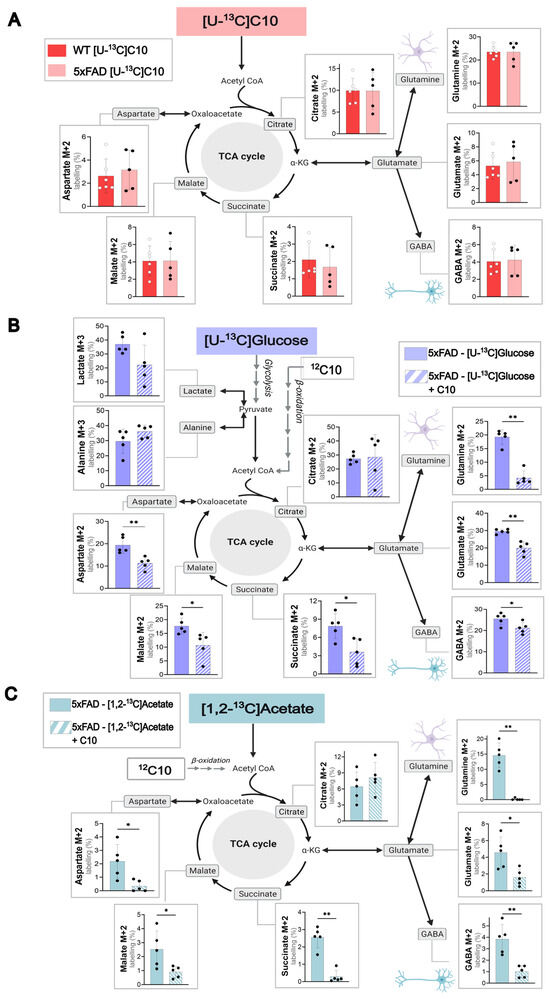
Figure 2.
C10 as a metabolic substrate in WT and 5xFAD mouse cerebral cortex slices. (A) metabolism of 0.2 mM [U-13C]C10 in acutely isolated cerebral cortical slices of WT and 5xFAD mice. (B) Metabolic competition assay between [U-13C]glucose and unlabeled 12C10 in cerebral cortical slices of 5xFAD mice. (C) Metabolic competition assay between [1,2-13C]acetate and unlabeled 12C10 in cerebral cortical slices of 5xFAD mice. Glycolytic products (M + 3) and first turn (M + 2) TCA cycle intermediates are shown. Concentrations: 5 mM glucose, 5 mM acetate, and 0.2 mM C10. Unless incubated with [U-13C]glucose, all incubations were supplemented with 5 mM D-glucose. Mean ± SD, n = 5–6 from individual animals, Welch’s t-test or Mann–Whitney test, * p < 0.05, ** p < 0.01.
To study the direct effect of C10 on glucose metabolism, brain slices from 5xFAD and WT mice were incubated with labeled [U-13C]glucose and unlabeled 12C10 in a competition assay. As the 13C10 enrichment levels in the tested metabolites (Figure 2A) were similar in WT and 5xFAD mice, the results below will focus on the 5xFAD mouse model (WT values in Figure S3B). Cortical slices incubated with [U-13C]glucose in the presence of unlabeled 12C10 showed a reduction of 13C-enrichment in the TCA cycle intermediates (succinate and malate) and the amino acids (glutamine, glutamate, GABA, and aspartate). The results indicate that C10 can compete with glucose as an energy substrate in the brain. By competing with glucose, the 12C carbons from C10 dilute the 13C-enrichment in the TCA cycle intermediates and amino acids from [U-13C]glucose. The significant decrease in 13C-enrichment was pronounced in the TCA cycle-derived amino acid glutamine M + 2 (p = <0.01). These findings correlated with the data in Figure 2A of the preferential metabolism of C10 by astrocytes. The same tendency was observed in WT and 5xFAD hippocampal slices metabolizing glucose (Figure S4).
As C10 can compete with glucose as a substrate for energy metabolism, we wanted to investigate whether C10 could influence acetate metabolism. Cortical brain slices of WT (Figure S3) and 5xFAD (Figure 2C) mice were incubated with labeled [1,2-13C]acetate and unlabeled 12C10. The labeling patterns for WT and 5xFAD mice were similar, so the following results focus on the 5xFAD mouse model. Figure 2C illustrates an overall decrease in 13C-enrichment derived from [1,2-13C]acetate when unlabeled C10 is present in the incubation medium. The most significant reduction in labeling (%) was seen in the TCA cycle intermediate succinate M + 2 (p = < 0.01) and the amino acid glutamine M + 2 (p = < 0.01). The same experiment was completed with hippocampal slices of WT and 5xFAD mice, which showed the same tendencies (Figure S4). The results strongly suggest that C10 can compete with acetate as an astrocyte energy substrate in cortical brain slices.
3.3. The Effect of C10 on Glucose Metabolism Is AD Mutation-Dependent in hiPSC-Derived Astrocytes
The incubation experiments in the 5xFAD brain slices showed that C10 could act as an energy substrate as well as affect glucose metabolism. These effects of C10 on cellular brain metabolism are most prominent in astrocytes; thus, we sought to determine whether C10 affects glucose metabolism in hiPSC-derived astrocytes. HiPSC-derived astrocytes with mutations in the amyloid precursor protein (APP) or presenilin-1 (PSEN-1) genes were used as a human cell model for familial forms of AD. HiPSC-derived astrocytes, K3P53 (referred to as WT astrocytes), and astrocytes carrying PSEN-1 A280E, APP London, or APP Swedish mutations (referred to as AD astrocytes) were incubated with [U-13C]glucose in the absence and presence of unlabeled 12C10 (Figure 3). When incubated with [U-13C]glucose in the presence of 12C10, WT astrocytes displayed increased 13C enrichment of α-ketoglutarate (p = 0.024) and succinate (p = 0.043) compared to enrichment from [U-13C]glucose alone. 13C enrichment for the remaining TCA cycle intermediates in the WT astrocytes was unaltered in response to 12C10. APP London astrocytes showed higher labeling in lactate (p = 0.049) and citrate ( p= 0.024) from [U-13C]glucose in the presence of 12C10 than from [U-13C]glucose. APP Swedish astrocytes incubated with [U-13C]glucose in the presence of 12C10 only resulted in increased labeling for α-ketoglutarate (p = 0.004). Interestingly, the presence of 12C10 slightly lowered glutamate labeling (p = 0.025) derived from [U-13C]glucose that was found in APP Swedish astrocytes. 13C enrichment for TCA cycle intermediates and TCA cycle-derived amino acids remained unchanged in PSEN-1 A280 astrocytes incubated in [U-13C]glucose media with or without 12C10. However, PSEN-1 A280E, along with WT and APP London astrocytes, showed lower labeling in alanine (p = 0.0079, p = 0.0067, p = 0.0039, respectively) when incubated with [U-13C]glucose in the presence of 12C10. Together, these results suggest that oxidative metabolism of glucose in these AD astrocytes remains mostly unchanged upon addition of C10, particularly in the PSEN-1 mutant.
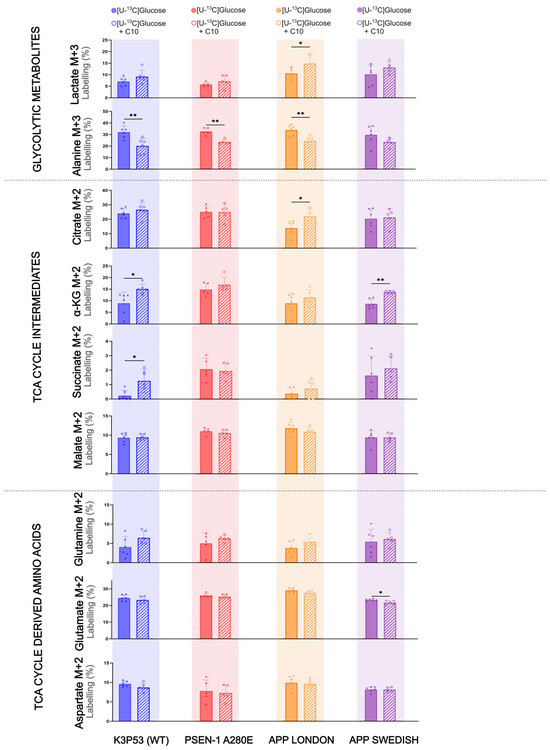
Figure 3.
The Influence of C10 on glucose metabolism in hiPSC-derived astrocytes carrying AD mutations. Metabolism of [U-13C]glucose in the presence and absence of 12C-C10 in hiPSC-derived astrocytes AD with a PSEN-1 or APP mutation, compared to WT. Glycolytic products (M + 3) and first turn (M + 2) TCA cycle intermediates are shown. Concentrations: 2.5 mM [U-13C]glucose or 2.5 mM [U-13C]glucose with 0.2 mM 12C-C10. Mean + SD, n = 3 independent differentiations; n = 5–6 technical replicates, Welch’s t-test or Mann–Whitney test, * p < 0.05, ** p < 0.01.
4. Discussion
In this study, we show that while glucose metabolism was largely maintained in both WT and 5xFAD cerebral cortical slices, acetate metabolism in 5xFAD cortices was lower compared to WT. Additionally, the MCFA C10 could act as an auxiliary brain energy substrate in both WT and 5xFAD. In hiPSC-derived AD astrocytes, there were distinct metabolic alterations in oxidative glucose metabolism upon C10 supplementation, though these responses varied across AD genotypes, emphasizing the complexity of astrocyte metabolic adaptations in AD.
4.1. Regional Changes in 5xFAD Brain Energy Metabolism
In our study, we did not observe a change in cortical [U-13C]glucose metabolism between WT and 5xFAD mice at 6 months of age (Figure 1A). There were, however, reductions in [U-13C] glucose-derived 13C- enrichment of TCA cycle intermediates in 5xFAD hippocampi, relative to WT (Figure S2). In a study specifically analyzing glucose uptake in the 5xFAD mouse model, despite increased Aβ burden in the brains at 2 and 5 months, it was only at 13 months that significant reductions to glucose metabolism could be seen in the whole brain and specific regions [36]. Another study looking at [18F]FDG utilization in 5xFAD mice found no change in uptake at 4 months; however, there was increased hippocampal [18F]FDG uptake at 8 and 12 months in 5xFAD compared to WT [37]. Hypermetabolic states have been reported in different brain regions, particularly in the early stages of human AD, furthering the need for more longitudinal studies into the glucose metabolism in different AD mouse models and how these compare to the human disease [38,39]. One of the reasons postulated for relatively preserved glucose uptake is microglial reconfiguration in the hippocampus, driving glucose uptake [37,40]. Also, because [18F]FDG uptake patterns on PET assess the hippocampus as a whole, the influence of different cell types cannot be distinguished. The use of labeled stable isotopes to map metabolism offers greater insight into oxidative metabolism in AD. In a previous study, we observed a hypometabolism of [U-13C]glucose in hippocampal slices and, to a lesser extent, cortical slices of 8-month-old 5xFAD mice [13] which agrees with what we see in the AD hippocampi of this study. Despite the 5xFAD model showing significant Aβ accumulation and gliosis from the age of 2 months [21] it is possible that with increased age, greater neurodegeneration exacerbates hypometabolism. Human studies demonstrating early changes to glucose metabolism also rely on relatively large sample sizes [41] A larger sample size in our study may have allowed detection of impaired glucose metabolism in our young 5xFAD mice.
The uptake of [1,2-13C]acetate, a substrate primarily metabolized in astrocytes, was used to investigate cell-specific metabolism [42]. In our study, we showed significantly lower labeling percentages in all metabolites of the TCA cycle and amino acids derived from [1,2-13C]acetate metabolism in 5xFAD cortices compared to WT (Figure 1B). This finding strongly indicates hampered astrocytic metabolism in the 6-month-old 5xFAD mice. This correlates with the astrogliosis found to occur in the 5xFAD model from as early as 2 months old [21] and previous studies where we have seen low 13C-enrichment of glutamine from [U-13C]glucose metabolism in 5xFAD mice [13,43]. Astrogliosis can occur as a response to Aβ pathology, as astrocytes near Aβ plaques become hypertrophic and proliferative [44], later leading to synaptic dysfunction and altered neurotransmission in AD [45,46,47]. Reactive astrocytes can become dysfunctional, with reduced ability to support neurons through metabolic coupling and neurotransmitter recycling [48]. From the findings in this study, it could be suggested that altered astrocytic metabolism might occur before glucose hypometabolism in the 5xFAD mouse model.
4.2. C10 as a Brain Substrate in AD and Its Effect on Glucose and Acetate Metabolism
Several studies have highlighted the benefits of MCFAs in neurological disorders, in particular MCFAs as brain energy substrates [16,49,50]. Through dynamic isotope labeling, we previously demonstrated that C10 is preferred over octanoic acid (C8) as a metabolic substrate in the cerebral cortex of NMRI mice and improved mitochondrial respiration [12]. The direct effect of C10 as a metabolite in an AD mouse model, and specifically the 5xFAD brain model, has remained relatively unexplored, so the next aim of our study was to first verify the extent to which C10 was metabolized under pathophysiological conditions. The use of [U-13C]C10 as an energy substrate was confirmed in brain slices of 5xFAD and WT mice (Figure 2A and Figure S3). Furthermore, no differences between WT and 5xFAD mice were observed in the 13C-enrichment of the TCA cycle intermediates and derived amino acids obtained from [U-13C]C10. This indicates that the two types of mice were equally effective in utilizing the MCFA as a brain energy substrate. These results agree with another study testing tridecanoin (the triglyceride of C10) in an epilepsy mouse model [17]. Tridecanoin was an anticonvulsant and improved mitochondrial function in the disease model. Though direct tracking of labeled MCFAs in the brain was not conducted, levels of C10 were elevated in the mice fed with the dietary supplement, suggesting a sustained ability to take up this substrate [17]. These findings are also in line with research suggesting that MCFAs could act as an alternative substrate during reduced glucose utilization, by bypassing the impaired enzymes associated with glycolysis [50,51,52]. In our study, the 13C-enrichment from [U-13C]C10 metabolism was highest in the amino acid glutamine with respect to other metabolites. This indicates that C10 is mainly metabolized in astrocytes, as glutamine is synthesized in astrocytes [23,53]. 13C-enrichment in the amino acid GABA via [U-13C]C10 metabolism was similar to what we have seen from [1,2-13C]acetate but much lower than enrichment from [U-13C]glucose (Figure 1 and Figure 2), which aligns with C10 being primarily metabolized in astrocytes. GABA is synthesized in GABAergic neurons via the use of astrocyte-derived glutamine, which is converted to glutamate via phosphate-activated glutaminase (PAG) and subsequently to GABA via glutamate decarboxylase (GAD). This astrocyte-derived glutamine is thought to be an essential substrate for neuronal GABA synthesis [54,55,56]; thus, the 13C-enrichment in GABA in our study likely comes from the C10-derived glutamine via the glutamate/GABA-glutamine cycle.
Our results also showed that unlabeled 12C10 reduced [U-13C]glucose metabolism in both WT and 5xFAD mice. This was detected by significantly lower 13C-enrichment in almost all metabolites from [U-13C]glucose metabolism when 12C10 was present in the incubation medium (Figure 2B). The same effect was seen when [1,2-13C]acetate was metabolized in the presence of 12C10. Though the majority of glucose in the brain is utilized by neurons for oxidative phosphorylation [57], astrocytes also metabolize glucose; hence, some of the competition between [U-13C]glucose and 12C10 could be attributed to astrocytes. It is also possible that neurons are able to metabolize C10 as demonstrated by the metabolism of C8 in hypothalamic neurons [58], however more research is needed in this area. Metabolic competition between [1,2-13C]acetate and 12C10 again substantiates the findings pointing to astrocytes as the main compartment for C10 metabolism. Competition between [1,2-13C]acetate and 12C10 was greater than between [U-13C]glucose and 12C10, characterized by a greater reduction in [1,2-13C]acetate labeling in the presence of C10 (Figure 2C).
Finally, our current metabolic findings may provide a mechanistic context for the functional effects of C10 on AMPA-mediated calcium signaling that we previously reported in hippocampal astrocytes (and neurons) from 5xFAD mice [20]. In that study, C10 exposure restored reduced AMPA-evoked calcium transients in both cell types to wild-type levels, suggesting improved excitatory signaling and cellular responsiveness. The present isotopic enrichment data indicate that C10 is readily oxidized in both wild-type and 5xFAD tissue and can partially compete with glucose and acetate for entry into central metabolic pathways. This efficient oxidative utilization of C10, particularly within astrocytic TCA cycle intermediates such as citrate and glutamine, supports the idea that C10 enhances metabolic flexibility and mitochondrial substrate availability. Such metabolic support may, in turn, facilitate the restoration of calcium handling and receptor function observed previously. Thus, when considered together, our functional and metabolic results suggest that C10 may modulate neuronal and astrocytic signaling roles, at least in part, by maintaining the energetic state of the tissue, enabling more robust receptor-mediated signaling in the AD context. However, further research is needed to determine how C10 affects cell-specific functional outcomes across different populations and pathological stages.
4.3. Effect of C10 on Glucose Metabolism in hiPSC Derived Astrocytes Carrying AD Mutations
Findings from the incubation experiments on hiPSC-derived astrocytes did not reveal differences in glucose metabolism between WT and AD mutants (Figure 3). Considering the reduction in acetate labeling in 5xFAD cortical slices compared to WT (Figure 1B). Other studies analyzing the metabolic activity of hiPSC PSEN, APP, or late onset AD (LOAD) mutant astrocytes via Seahorse have demonstrated increased oxidative consumption rates (OCR) and extracellular acidification rates (ECAR) compared to controls [29,59,60]. Coupled with increased ROS, oxidative stress, and reduced lactate production in AD astrocytes, this dysfunction could contribute to AD pathology [59]. While APP and PSEN1 mutations classically drive neuronal Aβ overproduction, accumulating evidence indicate that familial AD mutations can modulate astrocyte biology independently of Aβ secretion [29,61]. Together, this highlights the relevance of further modeling mutation-specific pathological changes in specific cellular populations. Our hiPSC-derived astrocyte model represents an early stage of AD. With extended maturation, these cells may develop additional metabolic dysfunction, as reported in studies using AD astrocytes matured for 7 weeks, and 5–8 months [59,62]. Mitochondrial stress tests also stimulate astrocytes, which could highlight metabolic differences compared to WT. Stimulation of astrocytes with inflammatory microglial factors such as IL-1α and TNFα increases glycolytic activity but not mitochondrial respiration [63]. The influence of C10 on glucose metabolism is less conclusive; our results indicate that while C10 partially modulated oxidative glucose metabolism in WT and certain AD-mutant astrocytes, they generally exhibited minimal metabolic adaptation to C10. Our observations that C10 increased 13C enrichment in certain TCA cycle intermediates and related metabolites in AD-mutant astrocytes are intriguing and suggest a more complex metabolic interaction. Rather than simply diluting the 13C label, C10 may stimulate glucose metabolism or hinder competing pathways, leading to increased flux of 13C from glucose, paradoxically increasing 13C enrichment into specific metabolic pools probing astrocytic metabolic compartmentalization. C10 may act as a metabolic modulator, enhancing mitochondrial function or altering substrate preference in a mutation-dependent manner. While hiPSC-derived AD astrocytes show no baseline glucose metabolism differences from WT under unstimulated conditions, metabolic alterations may arise with maturation or stress. C10 modulates glucose metabolism in a mutation-dependent way, highlighting metabolic heterogeneity among AD genotypes and underscoring the need for further investigation.
Astrocytic identity and reactivity are important considerations when interpreting metabolic phenotypes in both brain slices and hiPSC-derived astrocytes. We have previously characterized GFAP expression in the 5xFAD model, demonstrating robust and age-dependent astrogliosis in cortical and hippocampal regions [22], and we have likewise documented stable GFAP and S100β expression across hiPSC-derived astrocyte differentiation batches [29], which we also validated in the current cultures (Supplementary Figure S1). These earlier studies establish both the astrocytic purity of the cultures and the expected reactive profile in the 5xFAD model. As mentioned earlier, reactive astrocytes undergo well-described metabolic reprogramming that can influence substrate handling in AD models. Evidence shows that astrocytic reactivity is associated with increased glycolytic flux and reduced oxidative TCA cycle activity [64]. Reactive states also alter glutamate–glutamine cycling and anaplerosis, often lowering glutamine synthesis and modifying aspartate and citrate labeling patterns [13]. These mechanisms are consistent with the reduced [1,2-13C]acetate utilization observed in 5xFAD slices and may reflect gliosis-driven changes in astrocytic acetyl-CoA metabolism. In contrast, hiPSC-derived astrocytes, showing limited spontaneous reactivity, displayed only subtle, mutation-dependent metabolic shifts, suggesting that acute reactivity may exert a stronger influence on substrate handling than cell-autonomous effects of familial AD mutations. This highlights astrocytic reactivity as a potential modulator of MCFA and acetate metabolism.
Although our isotopic approach strongly indicates astrocytic involvement, particularly through glutamine labeling patterns, cortical slice experiments inherently include multiple viable cell types, including neurons and other types of glial cells. Therefore, the observed metabolic alterations in the 5xFAD tissue could also arise from indirect, non-cell-autonomous effects driven by incipient neuronal dysfunction or neuroinflammatory signaling. The reduced [1,2-13C]acetate metabolism in 5xFAD slices is consistent with impaired astrocytic function, yet we cannot exclude contributions from altered neuron–astrocyte metabolic coupling or microglial reprogramming. This contrasts with our isolated hiPSC-derived astrocyte data, where AD mutations alone only partially influence oxidative metabolism, suggesting that the in vivo disease microenvironment, including cellular cross-talk, inflammatory cues, and amyloid pathology, may be required to elicit the metabolic phenotype we detect in the 5xFAD model. Future studies employing astrocyte-specific metabolic reporters or cell-type-specific tracing strategies will be critical to dissect the relative contributions of each cell population to the metabolic changes observed.
In conclusion, using 5xFAD mice and human iPSC-derived astrocytes with familial AD mutations, our study found that C10 can compete with glucose and acetate as an energy substrate. Our findings suggest that C10 could act as an auxiliary substrate to help restore brain energy fitness in AD, highlighting its therapeutic potential.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells14242007/s1. Figure S1. Immunocytochemistry labelling of hiPSC-astrocytes; Figure S2. Glucose and acetate metabolism in WT and 5xFAD hippocampal brain slices; Figure S3. C10 as a metabolic substrate in mouse hippocampal and cortical slices; Figure S4. Metabolic competition assay between glucose and C10 in mouse hippocampal slices; Tables S1–S15. Full Statistical Report.
Author Contributions
Conceptualization, A.O.A., B.I.A., and K.B.; experimental design and work, A.O.A., B.I.A., M.B.R., and K.S.D.; writing, original draft preparation, A.O.A., M.B.R., K.K.F., K.B., and B.I.A.; review and editing A.O.A., and B.I.A. All authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was supported by The Independent Research Fund Denmark [grant number 1030-00285B] for AOA’s PhD stipend and consumables in BIA´s laboratory. KSD is supported by Innovation Fund Denmark [grant number 2078-00002B], and stem cell-related experiments were supported by the Alzheimerforeningen research grants awarded to BIA and KKF.
Institutional Review Board Statement
All animal experiments and procedures were approved by the Animal Welfare Committee (permit number: 2020-15-0201-00441) appointed by the Danish Ministry of Justice and in accordance with the European Communities Council Directive of 22 September 2010 (2010/63/EU) on the Protection of Animals Used for Experimental and Other Scientific Purposes.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Material. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors would like to acknowledge the excellent technical assistance of laboratory technician Heidi Marie Nielsen.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| 13C | Carbon-13 isotope |
| ACSF | Artificial cerebrospinal fluid |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| APOE | Apolipoprotein E |
| C10 | Decanoic acid |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FAD | Familial Alzheimer’s disease |
| GABA | Gamma-aminobutyric acid |
| GC–MS | Gas chromatography–mass spectrometry |
| hiPSC | Human induced pluripotent stem cell |
| MCI | Mild cognitive impairment |
| MCFA | Medium-chain fatty acid |
| MCT | Medium-chain triglyceride |
| PSEN1 | Presenilin 1 |
| SMAD | Small mothers against decapentaplegic |
| TCA | Tricarboxylic acid cycle |
References
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Frontzkowski, L.; Ewers, M.; Brendel, M.; Biel, D.; Ossenkoppele, R.; Hager, P.; Steward, A.; Dewenter, A.; Römer, S.; Rubinski, A.; et al. Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat. Commun. 2022, 13, 4899. [Google Scholar] [CrossRef]
- Patel, V.; Mill, J.; Okonkwo, O.C.; Salamat, S.; Li, L.; Raife, T. Global Energy Metabolism Deficit in Alzheimer Disease Brain. J. Prev. Alzheimer’s Dis. 2024, 11, 171–178. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Ameen, A.O.; Freude, K.; Aldana, B.I. Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease. Biomedicines 2022, 10, 2778. [Google Scholar] [CrossRef]
- Sun, L.; Ye, K.X.; Wong, H.L.K.; Wang, L.; Lim, S.L.; Chao, Y.X.; Zhang, C.; Yap, K.Z.; Feng, L. The Effects of Medium Chain Triglyceride for Alzheimer’s Disease Related Cognitive Impairment: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2023, 94, 441–456. [Google Scholar] [CrossRef]
- Giannos, P.; Prokopidis, K.; Lidoriki, I.; Triantafyllidis, K.K.; Kechagias, K.S.; Celoch, K.; Candow, D.G.; Ostojic, S.M.; Forbes, S.C. Medium-chain triglycerides may improve memory in non-demented older adults: A systematic review of randomized controlled trials. BMC Geriatr. 2022, 22, 817. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; E Turcotte, É.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef]
- Andersen, J.V.; Westi, E.W.; Neal, E.S.; Aldana, B.I.; Borges, K. β-Hydroxybutyrate and Medium-Chain Fatty Acids are Metabolized by Different Cell Types in Mouse Cerebral Cortex Slices. Neurochem. Res. 2023, 48, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Westi, E.W.; Jakobsen, E.; Urruticoechea, N.; Borges, K.; Aldana, B.I. Astrocyte metabolism of the medium-chain fatty acids octanoic acid and decanoic acid promotes GABA synthesis in neurons via elevated glutamine supply. Mol. Brain 2021, 14, 132. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Adamson, D.C. Neuronal-Astrocyte Metabolic Interactions: Understanding the Transition Into Abnormal Astrocytoma Metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Mett, J.; Müller, U. The medium-chain fatty acid decanoic acid reduces oxidative stress levels in neuroblastoma cells. Sci. Rep. 2021, 11, 6135. [Google Scholar] [CrossRef]
- Tan, K.N.; Carrasco-Pozo, C.; McDonald, T.S.; Puchowicz, M.; Borges, K. Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J. Cereb. Blood Flow Metab. 2017, 37, 2035–2048. [Google Scholar] [CrossRef]
- Warren, E.C.; Dooves, S.; Lugarà, E.; Damstra-Oddy, J.; Schaf, J.; Heine, V.M.; Walker, M.C.; Williams, R.S.B. Decanoic acid inhibits mTORC1 activity independent of glucose and insulin signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 23617–23625. [Google Scholar] [CrossRef]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- Abghari, M.; Vu, J.T.C.M.; Eckberg, N.; Aldana, B.I.; Kohlmeier, K.A. Decanoic Acid Rescues Differences in AMPA-Mediated Calcium Rises in Hippocampal CA1 Astrocytes and Neurons in the 5xFAD Mouse Model of Alzheimer’s Disease. Biomolecules 2023, 13, 1461. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129. [Google Scholar] [CrossRef]
- Westi, E.W.; Molhemi, S.; Hansen, C.T.; Skoven, C.S.; Knopper, R.W.; Ahmad, D.A.; Rindshøj, M.B.; Ameen, A.O.; Hansen, B.; Kohlmeier, K.A.; et al. Comprehensive Analysis of the 5xFAD Mouse Model of Alzheimer’s Disease Using dMRI, Immunohistochemistry, and Neuronal and Glial Functional Metabolic Mapping. Biomolecules 2024, 14, 1294. [Google Scholar] [CrossRef] [PubMed]
- McNair, L.F.; Kornfelt, R.; Walls, A.B.; Andersen, J.V.; Aldana, B.I.; Nissen, J.D.; Schousboe, A.; Waagepetersen, H.S. Metabolic Characterization of Acutely Isolated Hippocampal and Cerebral Cortical Slices Using [U-(13)C]Glucose and [1,2-(13)C]Acetate as Substrates. Neurochem. Res. 2017, 42, 810–826. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.R.; Holst, B.; Ramakrishna, S.; Muddashetty, R.; Schmid, B.; Freude, K. Generation of two iPSC lines with either a heterozygous V717I or a heterozygous KM670/671NL mutation in the APP gene. Stem Cell Res. 2019, 34, 101368. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.R.; Holst, B.; Mau-Holzmann, U.A.; Freude, K.; Schmid, B. Generation of two isogenic iPSC lines with either a heterozygous or a homozygous E280A mutation in the PSEN1 gene. Stem Cell Res. 2019, 35, 101403. [Google Scholar] [CrossRef]
- Dittlau, K.S.; Chandrasekaran, A.; Freude, K.; Van Den Bosch, L. Generation of Human Induced Pluripotent Stem Cell (hiPSC)-Derived Astrocytes for Amyotrophic Lateral Sclerosis and Other Neurodegenerative Disease Studies. Bio-Protocol 2024, 14, e4936. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Avci, H.X.; Ochalek, A.; Rösingh, L.N.; Molnár, K.; László, L.; Bellák, T.; Téglási, A.; Pesti, K.; Mike, A.; et al. Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Res. 2017, 25, 139–151. [Google Scholar] [CrossRef]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef]
- Salcedo, C.; Pozo Garcia, V.; García-Adán, B.; Ameen, A.O.; Gegelashvili, G.; Waagepetersen, H.S.; Freude, K.K.; Aldana, B.I. Increased glucose metabolism and impaired glutamate transport in human astrocytes are potential early triggers of abnormal extracellular glutamate accumulation in hiPSC-derived models of Alzheimer’s disease. J. Neurochem. 2023, 168, 822–840. [Google Scholar] [CrossRef]
- Wlaź, P.; Socała, K.; Nieoczym, D.; Żarnowski, T.; Żarnowska, I.; Czuczwar, S.J.; Gasior, M. Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 110–116. [Google Scholar] [CrossRef]
- Wlaź, P.; Socała, K.; Nieoczym, D.; Łuszczki, J.J.; Żarnowska, I.; Żarnowski, T.; Czuczwar, S.J.; Gasior, M. Anticonvulsant profile of caprylic acid, a main constituent of the medium-chain triglyceride (MCT) ketogenic diet, in mice. Neuropharmacology 2012, 62, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Bak, L.K.; Sonnewald, U.; Schousboe, A.; Waagepetersen, H.S. Metabolic Mapping of Astrocytes and Neurons in Culture Using Stable Isotopes and Gas Chromatography-Mass Spectrometry (GC-MS). In Brain Energy Metabolism; Hirrlinger, J., Waagepetersen, H.S., Eds.; Springer: New York, NY, USA, 2014; pp. 73–105. [Google Scholar]
- Sonnewald, U.; Westergaard, N.; Schousboe, A.; Svendsen, J.S.; Unsgård, G.; Petersen, S.B. Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem. Int. 1993, 22, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Schousboe, A. Glial Glutamine Homeostasis in Health and Disease. Neurochem. Res. 2023, 48, 1100–1128. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, N.; Waagepetersen, H.S.; Belhage, B.; Schousboe, A. Citrate, a Ubiquitous Key Metabolite with Regulatory Function in the CNS. Neurochem. Res. 2017, 42, 1583–1588. [Google Scholar] [CrossRef]
- Macdonald, I.R.; DeBay, D.R.; Reid, G.A.; O’Leary, T.P.; Jollymore, C.T.; Mawko, G.; Burrell, S.; Martin, E.; Bowen, C.V.; Brown, R.E.; et al. Early detection of cerebral glucose uptake changes in the 5XFAD mouse. Curr. Alzheimer Res. 2014, 11, 450–460. [Google Scholar] [CrossRef]
- Choi, H.; Choi, Y.; Lee, E.J.; Kim, H.; Lee, Y.; Kwon, S.; Hwang, D.W.; Lee, D.S. Hippocampal glucose uptake as a surrogate of metabolic change of microglia in Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 190. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef]
- Ashraf, A.; Fan, Z.; Brooks, D.J.; Edison, P. Cortical hypermetabolism in MCI subjects: A compensatory mechanism? Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 447–458. [Google Scholar] [CrossRef]
- Xiang, X.; Wind, K.; Wiedemann, T.; Blume, T.; Shi, Y.; Briel, N.; Beyer, L.; Biechele, G.; Eckenweber, F.; Zatcepin, A.; et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci. Transl. Med. 2021, 13, eabe5640. [Google Scholar] [CrossRef]
- Benzinger, T.L.S.; Blazey, T.; Jack, C.R.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.M.; Bateman, R.J.; et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–E4509. [Google Scholar] [CrossRef]
- Wyss, M.T.; Magistretti, P.J.; Buck, A.; Weber, B. Labeled acetate as a marker of astrocytic metabolism. J. Cereb. Blood Flow Metab. 2011, 31, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-Delcastillo, M.; Sun, R.C.; et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 2021, 12, 954. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef]
- Steele, M.L.; Robinson, S.R. Reactive astrocytes give neurons less support: Implications for Alzheimer’s disease. Neurobiol. Aging 2012, 33, 423.e1–423.e13. [Google Scholar] [CrossRef]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Micioni Di Bonaventura, M.V.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef]
- Khabbush, A.; Orford, M.; Tsai, Y.C.; Rutherford, T.; O’Donnell, M.; Eaton, S.; Heales, S.J.R. Neuronal decanoic acid oxidation is markedly lower than that of octanoic acid: A mechanistic insight into the medium-chain triglyceride ketogenic diet. Epilepsia 2017, 58, 1423–1429. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. BioMed Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Egan, J.M.; Mattson, M.P.; Kapogiannis, D. Medium Chain Triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Res. Rev. 2020, 58, 101001. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; McNair, L.F.; Schousboe, A.; Waagepetersen, H.S. Specificity of exogenous acetate and glutamate as astrocyte substrates examined in acute brain slices from female mice using methionine sulfoximine (MSO) to inhibit glutamine synthesis. J. Neurosci. Res. 2017, 95, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Leke, R.; Schousboe, A. The Glutamine Transporters and Their Role in the Glutamate/GABA-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 223–257. [Google Scholar] [CrossRef]
- Hertz, L. The Glutamate-Glutamine (GABA) Cycle: Importance of Late Postnatal Development and Potential Reciprocal Interactions between Biosynthesis and Degradation. Front. Endocrinol. 2013, 4, 59. [Google Scholar] [CrossRef]
- Lundgaard, I.; Li, B.; Xie, L.; Kang, H.; Sanggaard, S.; Haswell, J.D.R.; Sun, W.; Goldman, S.; Blekot, S.; Nielsen, M.; et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 2015, 6, 6807. [Google Scholar] [CrossRef]
- Haynes, V.R.; Michael, N.J.; van den Top, M.; Zhao, F.Y.; Brown, R.D.; De Souza, D.; Dodd, G.T.; Spanswick, D.; Watt, M.J. A Neural basis for Octanoic acid regulation of energy balance. Mol. Metab. 2020, 34, 54–71. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Ryu, W.-I.; Bormann, M.K.; Shen, M.; Kim, D.; Forester, B.; Park, Y.; So, J.; Seo, H.; Sonntag, K.-C.; Cohen, B.M. Brain cells derived from Alzheimer’s disease patients have multiple specific innate abnormalities in energy metabolism. Mol. Psychiatry 2021, 26, 5702–5714. [Google Scholar] [CrossRef]
- Ziff, O.J.; Parfitt, G.M.; Jolly, S.; Casey, J.M.; Granat, L.; Samra, S.; Setó-Salvia, N.; Alatza, A.; Phadke, L.; Galet, B.; et al. Mutations in PSEN1 predispose inflammation in an astrocyte model of familial Alzheimer’s disease through disrupted regulated intramembrane proteolysis. Mol. Neurodegener. 2025, 20, 73. [Google Scholar] [CrossRef]
- Konttinen, H.; Gureviciene, I.; Oksanen, M.; Grubman, A.; Loppi, S.; Huuskonen, M.T.; Korhonen, P.; Lampinen, R.; Keuters, M.; Belaya, I.; et al. PPARβ/δ-agonist GW0742 ameliorates dysfunction in fatty acid oxidation in PSEN1ΔE9 astrocytes. Glia 2019, 67, 146–159. [Google Scholar] [CrossRef]
- McComish, S.F.; O’Sullivan, J.; Copas, A.M.M.; Imiolek, M.; Boyle, N.T.; Crompton, L.A.; Lane, J.D.; Caldwell, M.A. Reactive astrocytes generated from human iPSC are pro-inflammatory and display altered metabolism. Exp. Neurol. 2024, 382, 114979. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, M.H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron–Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).