
Cellular and Molecular Mechanisms Explaining the Link Between Inflammatory Bowel Disease and Heart Failure



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
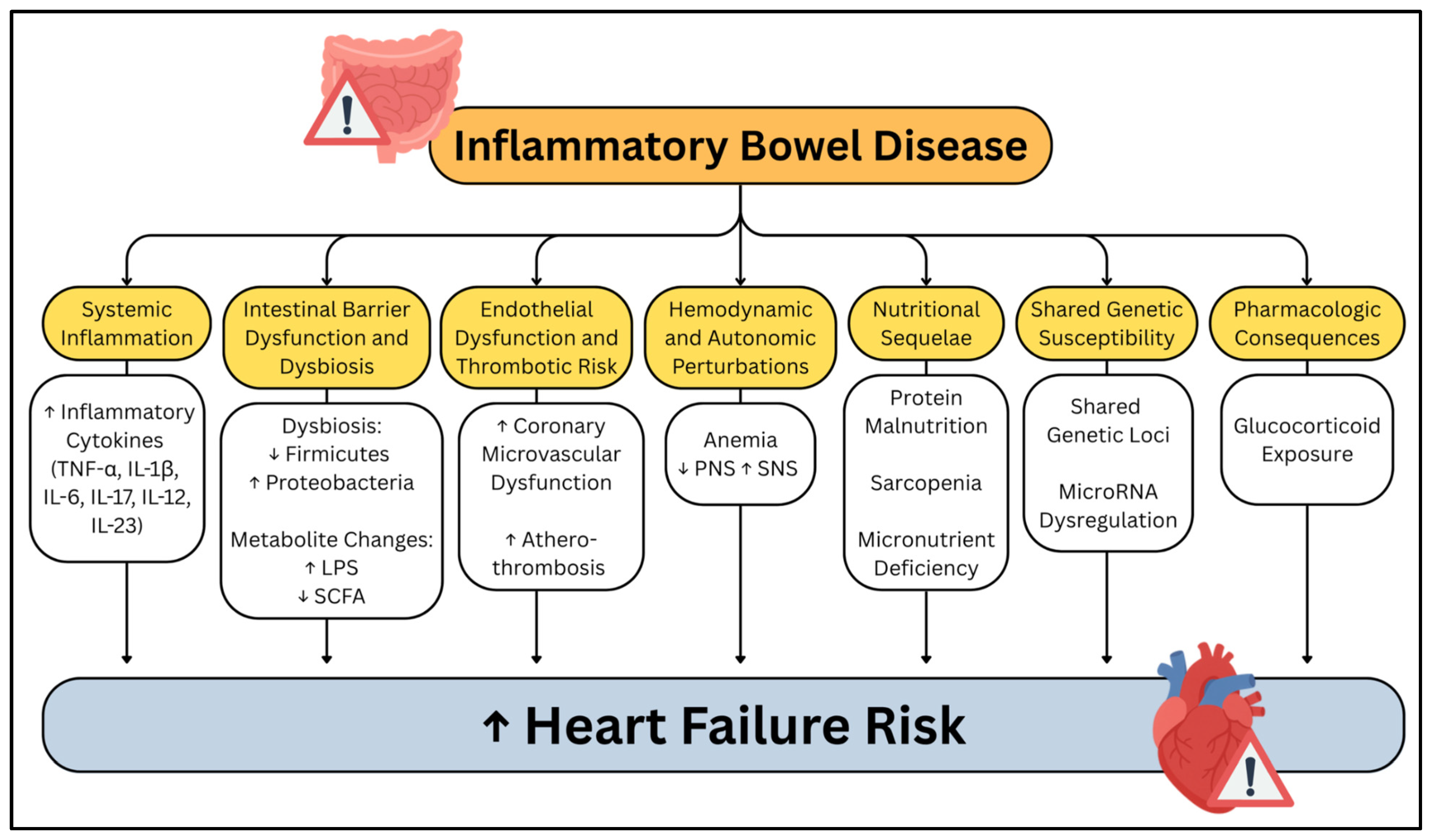
3. Systemic Inflammation
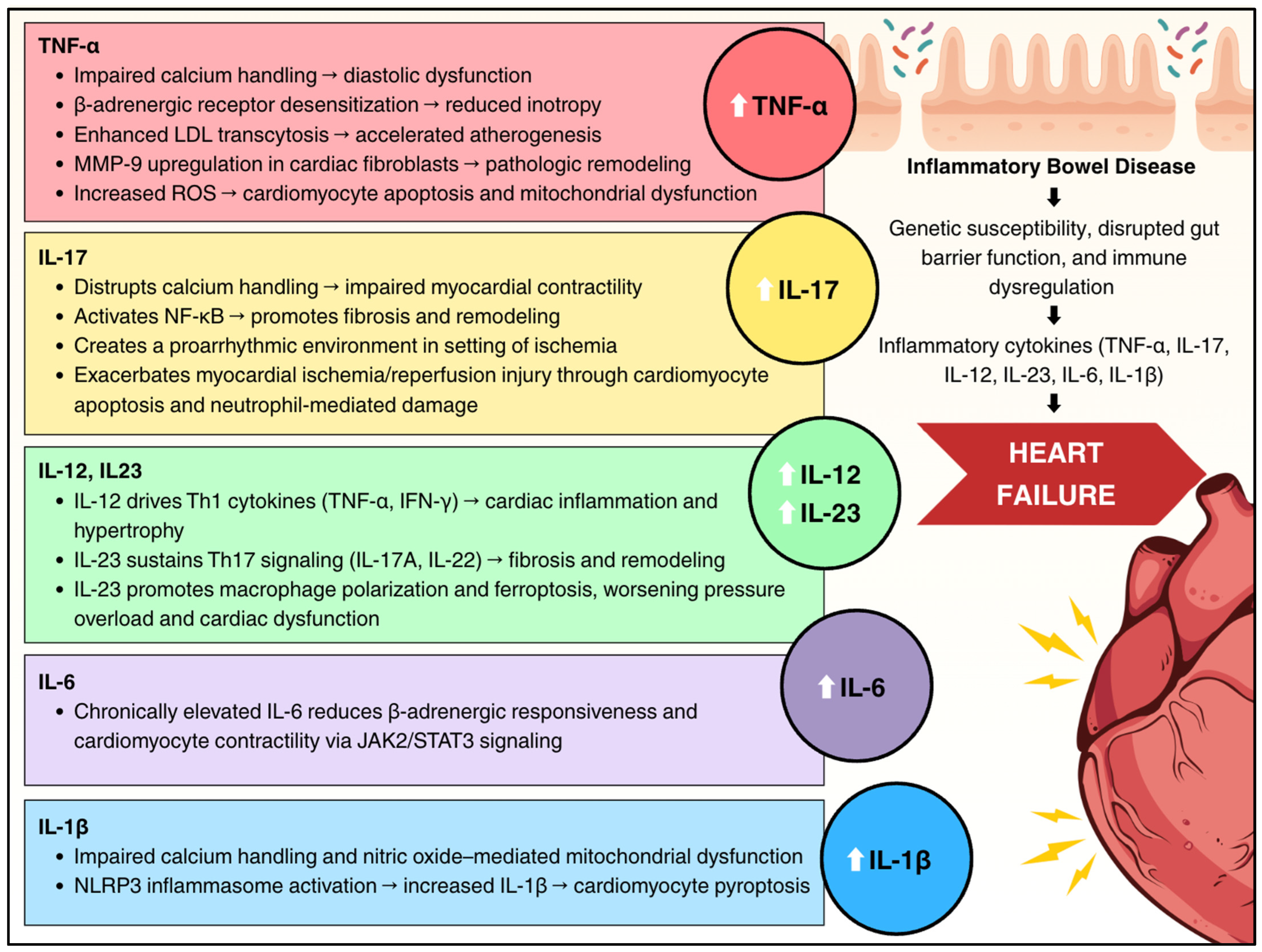
3.1. Cytokine-Mediated Myocardial Dysfunction
3.2. Tumor Necrosis Factor-Alpha
3.3. Interleukin-1β
3.4. Interleukin-6
3.5. Interleukin-12 and Interleukin-23
3.6. Interleukin-17
3.7. Interleukin-33
4. Experimental Evidence
Intestinal Barrier Dysfunction and Gut Microbiome Alteration
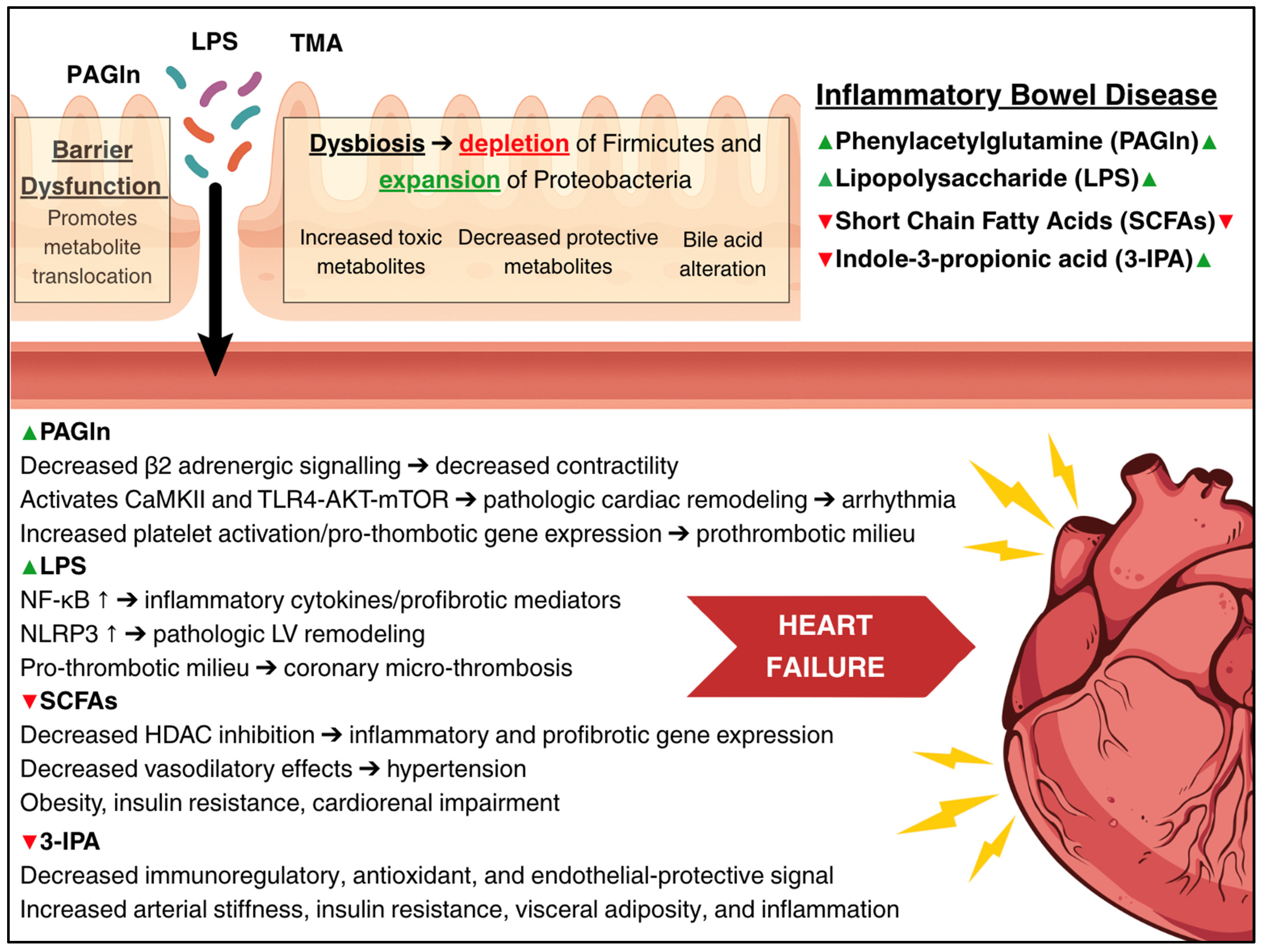
5. Intestinal Barrier Dysfunction
5.1. Structural and Functional Changes
5.2. Cardiovascular Implications
5.3. Lipopolysaccharide and Cardiac Dysfunction
5.4. Does TMAO Play a Role?
6. Gut Microbiome Dysbiosis
6.1. Microbial Composition Changes
6.2. Short-Chain Fatty Acid Depletion
6.3. Do Bile Acids Play a Role?
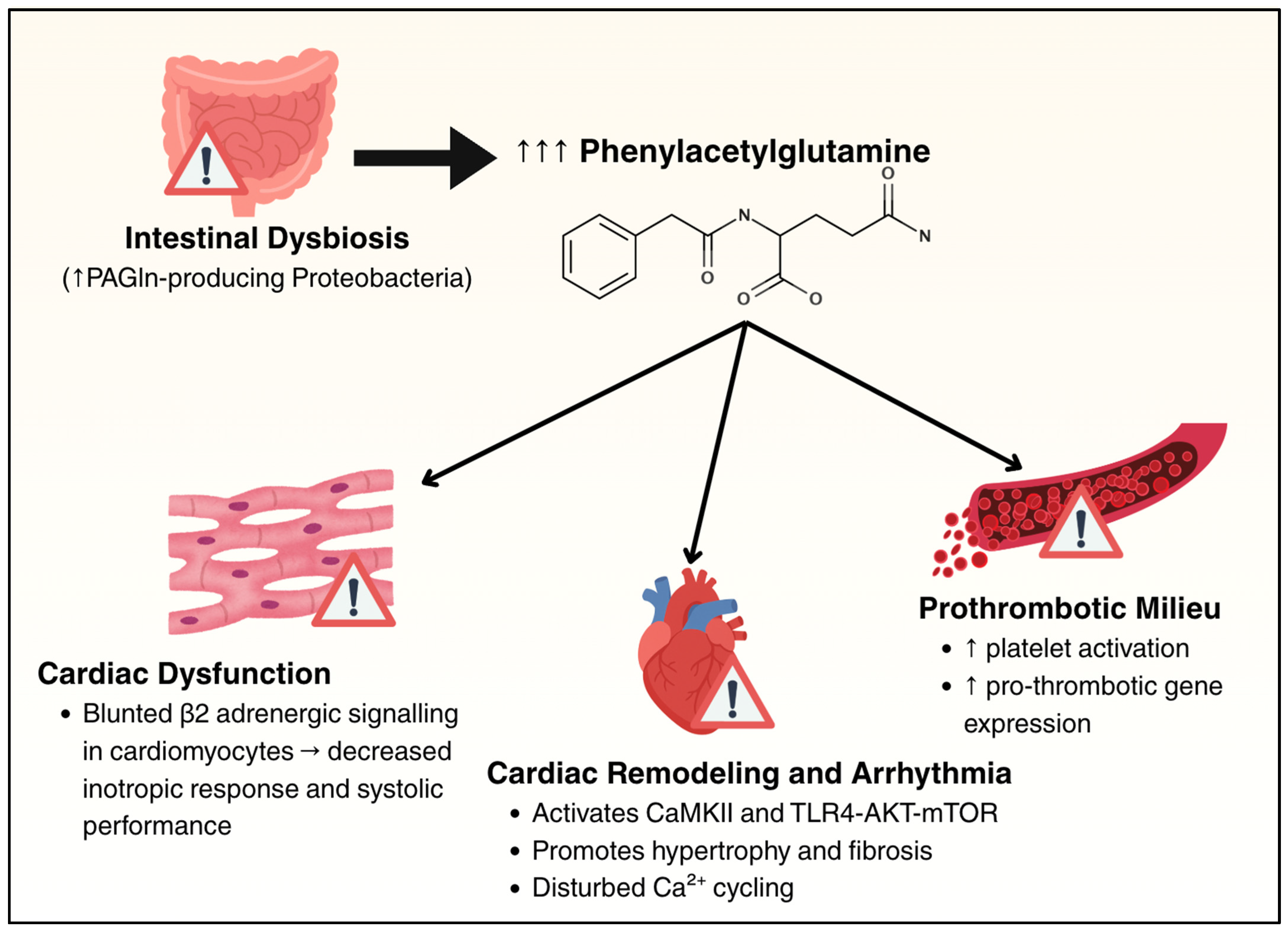
6.4. Phenylacetylglutamine
6.5. Tryptophan-Derived Indoles
6.6. Summary of Dysbiosis and Barrier Dysfunction
7. Endothelial and Vascular Dysfunction and Thrombotic Risk
7.1. Systemic Endothelial Dysfunction
7.2. Coronary Microvascular Dysfunction
7.3. Accelerated Atherosclerosis and Thrombotic Risk
8. Hemodynamic and Autonomic Perturbations
8.1. Hemodynamic Changes
8.2. Autonomic Nervous System Dysfunction
9. Nutritional Sequelae
9.1. Protein Malnutrition and Sarcopenia
9.2. Micronutrient Deficiencies
10. Shared Genetic and Epigenetic Susceptibility
10.1. Shared Genetic Loci
10.2. MicroRNA Dysregulation
10.3. Pharmacology
10.4. Clinical Implications
11. Future Directions
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olpin, J.D.; Sjoberg, B.P.; Stilwill, S.E.; Jensen, L.E.; Rezvani, M.; Shaaban, A.M. Beyond the Bowel: Extraintestinal Manifestations of Inflammatory Bowel Disease. RadioGraphics 2017, 37, 1135–1160. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Singh, A.; Kavanaugh, A.; Rubin, D.T. Extraintestinal Manifestations of Inflammatory Bowel Disease: Current Concepts, Treatment, and Implications for Disease Management. Gastroenterology 2021, 161, 1118–1132. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Rohit Reddy, S.; Llukmani, A.; Hashim, A.; Haddad, D.R.; Patel, D.S.; Ahmad, F.; Gordon, D.K. Cardiovascular Manifestations in Inflammatory Bowel Disease: A Systematic Review of the Pathogenesis and Management of Pericarditis. Cureus 2021, 13, e14010. [Google Scholar] [CrossRef] [PubMed]
- Aniwan, S.; Pardi, D.S.; Tremaine, W.J.; Loftus, E.V. Increased Risk of Acute Myocardial Infarction and Heart Failure in Patients With Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2018, 16, 1607–1615.e1. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yao, J.; Olén, O.; Halfvarson, J.; Bergman, D.; Ebrahimi, F.; Rosengren, A.; Sundström, J.; Ludvigsson, J.F. Risk of Heart Failure in Inflammatory Bowel Disease: A Swedish Population-Based Study. Eur. Heart J. 2024, 45, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, S.L.; Ahlehoff, O.; Lindhardsen, J.; Erichsen, R.; Lamberts, M.; Khalid, U.; Nielsen, O.H.; Torp-Pedersen, C.; Gislason, G.H.; Hansen, P.R. Inflammatory Bowel Disease Is Associated with an Increased Risk of Hospitalization for Heart Failure: A Danish Nationwide Cohort Study. Circ. Heart Fail. 2014, 7, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.A.; Fiuza, J.G.; Rodrigues, C.A.M.; Craveiro, N.; Gil Pereira, J.; Sousa, P.C.R.F.; Martins, D.C.P.; Cancela, E.M.; Ministro Dos Santos, M.P. Inflammatory Bowel Disease and Cardiac Function: A Systematic Review of Literature with Meta-Analysis. Ther. Adv. Gastroenterol. 2024, 17, 17562848241299534. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-T.; Wu, C.-K.; Lee, J.-K.; Chang, S.-N.; Kuo, Y.-M.; Wang, Y.-C.; Lai, L.-P.; Chiang, F.-T.; Hwang, J.-J.; Lin, J.-L. TNF-down-Regulates Sarcoplasmic Reticulum Ca2+ ATPase Expression and Leads to Left Ventricular Diastolic Dysfunction through Binding of NF-B to Promoter Response Element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kline, K.T.; Zhong, X.S.; Xiao, Y.; Lian, H.; Peng, J.; Liu, X.; Powell, D.W.; Tang, G.; Li, Q. Chronic Colitis Upregulates microRNAs Suppressing Brain-Derived Neurotrophic Factor in the Adult Heart. PLoS ONE 2021, 16, e0257280. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.T.; Mohan, M.L.; Gupta, M.K.; Martelli, E.E.; Hussain, A.K.; Qin, Y.; Chandrasekharan, U.M.; Young, D.; Feldman, A.M.; Sen, S.; et al. Gβγ-Independent Recruitment of G-Protein Coupled Receptor Kinase 2 Drives Tumor Necrosis Factor α–Induced Cardiac β-Adrenergic Receptor Dysfunction. Circulation 2013, 128, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Andresen, B.; Hill, M.; Zhang, J.; Booth, F.; Zhang, C. Role of Reactive Oxygen Species in Tumor Necrosis Factor-Alpha Induced Endothelial Dysfunction. Curr. Hypertens. Rev. 2008, 4, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xue, F.; Cheng, C.; Sui, W.; Zhang, J.; Meng, L.; Lu, Y.; Xiong, W.; Bu, P.; Xu, F.; et al. Cardiomyocyte-Specific Knockout of ADAM17 Alleviates Doxorubicin-Induced Cardiomyopathy via Inhibiting TNFα–TRAF3–TAK1–MAPK Axis. Signal Transduct. Target. Ther. 2024, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, N.; Soorappan, R.N.; Haque, M.; Sriramula, S.; Francis, J. TNF-α-Induced Mitochondrial Oxidative Stress and Cardiac Dysfunction: Restoration by Superoxide Dismutase Mimetic Tempol. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H2726–H2737. [Google Scholar] [CrossRef] [PubMed]
- Rolski, F.; Błyszczuk, P. Complexity of TNF-α Signaling in Heart Disease. J. Clin. Med. 2020, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.E.; Pontes, M.H.B.; Cantão, M.B.S.; Prado, A.F. The Role of Matrix Metalloproteinase-9 in Cardiac Remodeling and Dysfunction and as a Possible Blood Biomarker in Heart Failure. Pharmacol. Res. 2024, 206, 107285. [Google Scholar] [CrossRef] [PubMed]
- Zhu, E.; Yuan, C.; Hu, S.; Liao, Y.; Li, B.; Zhou, Y.; Zhou, W. Injection of Matrix Metalloproteinase-9 Leads to Ventricular Remodeling. Dis. Markers 2022, 2022, 1659771. [Google Scholar] [CrossRef] [PubMed]
- Szekely, Y.; Arbel, Y. A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today? Cardiol. Ther. 2018, 7, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The Importance of NLRP3 Inflammasome in Heart Failure. J. Card. Fail. 2015, 21, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Neurath, M.F. IL-6 Signaling in Inflammatory Bowel Disease: Pathophysiological Role and Clinical Relevance: Inflamm. Bowel Dis. 2007, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.Y.; Rabkin, S.W. C-Reactive Protein, Interleukin-6, Trimethylamine-N-Oxide, Syndecan-1, Nitric Oxide, and Tumor Necrosis Factor Receptor-1 in Heart Failure with Preserved Versus Reduced Ejection Fraction: A Meta-Analysis. Curr. Heart Fail. Rep. 2023, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chia, Y.C.; Kieneker, L.M.; Van Hassel, G.; Binnenmars, S.H.; Nolte, I.M.; Van Zanden, J.J.; Van Der Meer, P.; Navis, G.; Voors, A.A.; Bakker, S.J.L.; et al. Interleukin 6 and Development of Heart Failure with Preserved Ejection Fraction in the General Population. J. Am. Heart Assoc. 2021, 10, e018549. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.F.; McDowell, K.; Welsh, P.; Petrie, M.C.; Anand, I.; Berg, D.D.; De Boer, R.A.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; et al. Interleukin-6 in Heart Failure with Reduced Ejection Fraction and the Effect of Dapagliflozin. JACC Heart Fail. 2025, 13, 102393. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.A.; Rose, N.R.; Čiháková, D. The Varying Faces of IL-6: From Cardiac Protection to Cardiac Failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yang, J.; Li, T.; Wang, X.; Fan, Z.; Ye, Q.; Du, Y. JAK/STAT3 Signaling in Cardiac Fibrosis: A Promising Therapeutic Target. Front. Pharmacol. 2024, 15, 1336102. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Salas, A.; Sands, B.E.; Abraham, C.; Leibovitzh, H.; Neurath, M.F.; Vande Casteele, N. Alimentiv Translational Research Consortium (ATRC) IL-12 and IL-23 Pathway Inhibition in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Berrebi, D.; Besnard, M.; Fromont-Hankard, G.; Paris, R.; Mougenot, J.F.; De Lagausie, P.; Emilie, D.; Cezard, J.P.; Navarro, J.; Peuchmaur, M. Interleukin-12 Expression Is Focally Enhanced in the Gastric Mucosa of Pediatric Patients with Crohn’s Disease. Am. J. Pathol. 1998, 152, 667–672. [Google Scholar] [PubMed]
- Trinchieri, G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Korta, A.; Kula, J.; Gomułka, K. The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 10172. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 Cytokines: From Discovery to Targeted Therapies for Immune-Mediated Inflammatory Diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, T.; Bot, I.; Kuiper, J. The IL-12 Cytokine Family in Cardiovascular Diseases. Cytokine 2019, 122, 154188. [Google Scholar] [CrossRef] [PubMed]
- Papamichail, A.; Kourek, C.; Briasoulis, A.; Xanthopoulos, A.; Tsougos, E.; Farmakis, D.; Paraskevaidis, I. Targeting Key Inflammatory Mechanisms Underlying Heart Failure: A Comprehensive Review. Int. J. Mol. Sci. 2023, 25, 510. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, U.; He, X.; Xu, R.; Liu, X.; Pan, L.; Sun, Y.; Chen, J.-X.; Chen, Y. IL-12α Deficiency Attenuates Pressure Overload-Induced Cardiac Inflammation, Hypertrophy, Dysfunction, and Heart Failure Progression. Front. Immunol. 2023, 14, 1105664. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ji, Q.; Pan, H.; Feng, Y.; Ye, D.; Gan, L.; Wan, J.; Ye, J. IL-23p19 Deficiency Reduces M1 Macrophage Polarization and Improves Stress-Induced Cardiac Remodeling by Alleviating Macrophage Ferroptosis in Mice. Biochem. Pharmacol. 2024, 222, 116072. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, H.; Tian, D.; Wang, G. TH17 Cell: A Double-Edged Sword in the Development of Inflammatory Bowel Disease. Ther. Adv. Gastroenterol. 2024, 17, 17562848241230896. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jia, X.; Xu, L.; Chen, H.; Xie, S.; Cai, J. Interleukin-17 and Inflammatory Bowel Disease: A 2-Sample Mendelian Randomization Study. Front. Immunol. 2023, 14, 1238457. [Google Scholar] [CrossRef] [PubMed]
- Sandip, C.; Tan, L.; Huang, J.; Li, Q.; Ni, L.; Cianflone, K.; Wang, D.W. Common Variants in IL-17A/IL-17RA Axis Contribute to Predisposition to and Progression of Congestive Heart Failure. Medicine 2016, 95, e4105. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Li, D.; Wang, Z.; Liu, Y.; Yang, J.; Li, C.; Li, X.; Ma, J.; Zhang, M.; Lu, Y.; et al. Interleukin-17 Upregulation Participates in the Pathogenesis of Heart Failure in Mice via NF-κB-Dependent Suppression of SERCA2a and Cav1.2 Expression. Acta Pharmacol. Sin. 2021, 42, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-L.; Hsiao, Y.-W.; Tsai, Y.-N.; Lin, S.-F.; Liu, S.-H.; Lin, Y.-J.; Lo, L.-W.; Chung, F.-P.; Chao, T.-F.; Hu, Y.-F.; et al. Interleukin-17 Enhances Cardiac Ventricular Remodeling via Activating MAPK Pathway in Ischemic Heart Failure. J. Mol. Cell. Cardiol. 2018, 122, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-H.; Xia, N.; Zhou, S.-F.; Tang, T.-T.; Yan, X.-X.; Lv, B.-J.; Nie, S.-F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17A Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Cardiomyocyte Apoptosis and Neutrophil Infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, M.; Watanabe, T.; Kamata, K.; Minaga, K.; Kudo, M. IL-33 as a Critical Cytokine for Inflammation and Fibrosis in Inflammatory Bowel Diseases and Pancreatitis. Front. Physiol. 2021, 12, 781012. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.-P. IL-33: An Alarmin Cytokine with Crucial Roles in Innate Immunity, Inflammation and Allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 Pathway: Therapeutic Target and Novel Biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Veeraveedu, P.T.; Sanada, S.; Okuda, K.; Fu, H.Y.; Matsuzaki, T.; Araki, R.; Yamato, M.; Yasuda, K.; Sakata, Y.; Yoshimoto, T.; et al. Ablation of IL-33 Gene Exacerbate Myocardial Remodeling in Mice with Heart Failure Induced by Mechanical Stress. Biochem. Pharmacol. 2017, 138, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Altara, R.; Ghali, R.; Mallat, Z.; Cataliotti, A.; Booz, G.W.; Zouein, F.A. Conflicting Vascular and Metabolic Impact of the IL-33/sST2 Axis. Cardiovasc. Res. 2018, 114, 1578–1594. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.H.; Che, X.; Kwak, M.S.; Kim, S.; Kim, J.H.; Ma, H.W.; Kim, D.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; et al. Interleukin-33 Regulates Intestinal Inflammation by Modulating Macrophages in Inflammatory Bowel Disease. Sci. Rep. 2017, 7, 851. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.S.; Lopez, K.M.; Krishnachaitanya, S.S.; Liu, M.; Xiao, Y.; Ou, R.; Nagy, H.I.; Kochkarian, T.; Powell, D.W.; Fujise, K.; et al. Fecal Microbiota Transplantation Mitigates Cardiac Remodeling and Functional Impairment in Mice with Chronic Colitis. bioRxiv 2025. [Google Scholar] [CrossRef] [PubMed]
- Nouraei, H.; Rabkin, S.W. A New Approach to the Clinical Subclassification of Heart Failure with Preserved Ejection Fraction. Int. J. Cardiol. 2021, 331, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Hemker, M.; Kahn, S.A.; Steinbach, W.J.; Section on Gastroenteroloy, Hepatology, and Nutrition; Cohen, M.B.; Brumbaugh, D.; Cole, C.; Dotson, J.L.; Harpavat, S.; Lightdale, J.R.; et al. Fecal Microbiota Transplantation: Information for the Pediatrician. Pediatrics 2023, 152, e2023062922. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.V.A.; Hwangbo, H.; Lai, Y.; Hong, S.B.; Choi, Y.-J.; Park, H.-J.; Ban, K. The Gut–Heart Axis: Updated Review for The Roles of Microbiome in Cardiovascular Health. Korean Circ. J. 2023, 53, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Kiritkumar Makwana, R.; Shetty, V.; Mukherjee, S.; Narayan, P. Cardiovascular Diseases and the Heart–Gut Cross Talk. Indian Heart J. 2024, 76, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Fromm, M.; Schulzke, J.-D. Determinants of Colonic Barrier Function in Inflammatory Bowel Disease and Potential Therapeutics. J. Physiol. 2012, 590, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in Expression and Distribution of Claudin 2, 5 and 8 Lead to Discontinuous Tight Junctions and Barrier Dysfunction in Active Crohn’s Disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.Y.; Söderholm, J.D. Importance of Disrupted Intestinal Barrier in Inflammatory Bowel Diseases: Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, L.A.C.; Matiollo, C.; Rosa, J.S.d.; Felisberto, M.; Dalmarco, E.M.; Schiavon, L.d.L. Factors associated with Circulating Zonulin in Inflammatory Bowel Disease. Arq. Gastroenterol. 2022, 59, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Gerova, V.A.; Stoynov, S.G.; Katsarov, D.S.; Svinarov, D.A. Increased Intestinal Permeability in Inflammatory Bowel Diseases Assessed by Iohexol Test. World J. Gastroenterol. 2011, 17, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.-H.; Wang, Y.; Zhang, X.-M.; Wang, W.-X.; Zhang, Q.; Tang, Y.-P.; Yue, S.-J. Intestinal Permeability in Human Cardiovascular Diseases: A Systematic Review and Meta-Analysis. Front. Nutr. 2024, 11, 1361126. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary Metabolism, the Gut Microbiome, and Heart Failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Cruz, C.; Rojas Huerta, A.; Lima Barrientos, J.; Rodriguez, C.; Devani, A.; Boosahda, V.; Rasagna Mareddy, N.S.; Briceno Silva, G.; Del Castillo Miranda, J.C.; Reyes Gochi, K.A.; et al. Inflammatory Bowel Disease and Cardiovascular Disease: An Integrative Review with a Focus on the Gut Microbiome. Cureus 2024, 16, e65136. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef] [PubMed]
- Rojo, Ó.P.; Román, A.L.S.; Arbizu, E.A.; De La Hera Martínez, A.; Sevillano, E.R.; Martínez, A.A. Serum Lipopolysaccharide-Binding Protein in Endotoxemic Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, F.; Bargi, R.; Hosseini, M.; Farzadnia, M.; Khazaei, M. Cardiac and Renal Fibrosis and Oxidative Stress Balance in Lipopolysaccharide-Induced Inflammation in Male Rats. ARYA Atheroscler. 2018, 14, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, X.; Liu, X.-T.; Li, X.-Y.; Zhu, H.-Z.; Xie, Y.-H.; Wang, S.-W.; Li, Y.; Zhao, Y. Carvacrol Alleviates LPS-Induced Myocardial Dysfunction by Inhibiting the TLR4/MyD88/NF-κB and NLRP3 Inflammasome in Cardiomyocytes. J. Inflamm. 2024, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Maluleke, T.T.; Manilall, A.; Shezi, N.; Baijnath, S.; Millen, A.M.E. Acute Exposure to LPS Induces Cardiac Dysfunction via the Activation of the NLRP3 Inflammasome. Sci. Rep. 2024, 14, 24378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Lira, A.L.; Taskin, B.; Puy, C.; Keshari, R.S.; Silasi, R.; Pang, J.; Aslan, J.E.; Shatzel, J.J.; Lorentz, C.U.; Tucker, E.I.; et al. The Physicochemical Properties of Lipopolysaccharide Chemotypes Regulate Activation of the Contact Pathway of Blood Coagulation. J. Biol. Chem. 2025, 301, 108110. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide Potentiates Platelet Responses via Toll-like Receptor 4-Stimulated Akt-Erk-PLA2 Signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Lipopolysaccharide Augments Venous and Arterial Thrombosis in the Mouse. Thromb. Res. 2008, 123, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.A.P.D.; Chaves, L.D.S.; Tarso Facundo, H. Mitochondrial Electron Transport Chain Disruption and Oxidative Stress in Lipopolysaccharide-Induced Cardiac Dysfunction in Rats and Mice. Free Radic. Res. 2025, 59, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Sandek, A.; Bjarnason, I.; Volk, H.-D.; Crane, R.; Meddings, J.B.; Niebauer, J.; Kalra, P.R.; Buhner, S.; Herrmann, R.; Springer, J.; et al. Studies on Bacterial Endotoxin and Intestinal Absorption Function in Patients with Chronic Heart Failure. Int. J. Cardiol. 2012, 157, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Gautier, T.; Masson, D.; Bouhemad, B.; Guinot, P.-G. Endotoxemia in Acute Heart Failure and Cardiogenic Shock: Evidence, Mechanisms and Therapeutic Options. J. Clin. Med. 2023, 12, 2579. [Google Scholar] [CrossRef] [PubMed]
- Kochkarian, T.; Nagy, H.I.; Li, Q. Gut–Heart Axis: Cardiac Remodeling and Heart Failure in the Context of Inflammatory Bowel Disease and Dysbiosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2025, 329, G122–G137. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z.; Cui, J.; Li, D.; Lu, J.; Cui, X.; Xie, L.; Wu, Y.; Lin, Q.; Li, Y. Trimethylamine N-Oxide in Heart Failure: A Meta-Analysis of Prognostic Value. Front. Cardiovasc. Med. 2022, 9, 817396. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Lemaitre, R.N.; Jensen, P.N.; Wang, M.; Wang, Z.; Li, X.S.; Nemet, I.; Lee, Y.; Lai, H.T.M.; De Oliveira Otto, M.C.; et al. Trimethylamine N-Oxide and Related Gut Microbe-Derived Metabolites and Incident Heart Failure Development in Community-Based Populations. Circ. Heart Fail. 2024, 17, e011569. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, Z.; Yan, J.; Liu, H.; Liu, Q.; Deng, Y.; Ou, C.; Chen, M. Gut Microbe-Derived Metabolite Trimethylamine N-Oxide Induces Cardiac Hypertrophy and Fibrosis. Lab. Investig. 2019, 99, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Feng, Y.; Wang, F.; Gao, L.; Wang, Y. Trimethylamine Oxide Promotes Myocardial Fibrosis through Activating JAK2-STAT3 Pathway. Biochem. Biophys. Res. Commun. 2025, 750, 151390. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Shimizu, I.; Shimada, A.; Nakahara, K.; Yanagisawa, S.; Kubo, M.; Fukuda, S.; Ishii, C.; Yamamoto, H.; Ishikawa, T.; et al. Brown Adipose Tissue Dysfunction Promotes Heart Failure via a Trimethylamine N-Oxide-Dependent Mechanism. Sci. Rep. 2022, 12, 14883. [Google Scholar] [CrossRef] [PubMed]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; VanDongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Teft, W.A.; Morse, B.L.; Choi, Y.-H.; Woolsey, S.; DeGorter, M.K.; Hegele, R.A.; Tirona, R.G.; Kim, R.B. Trimethylamine-N-Oxide: A Novel Biomarker for the Identification of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Laryushina, Y.; Samoilova-Bedych, N.; Turgunova, L.; Marchenko, A.; Turgunov, Y. Association of TMAO Levels with Indicators of Ulcerative Colitis Activity. Casp. J. Intern. Med. 2025, 16, 114. [Google Scholar] [CrossRef]
- Laryushina, Y.; Samoilova-Bedych, N.; Turgunova, L.; Kozhakhmetov, S.; Alina, A.; Suieubayev, M.; Mukhanbetzhanov, N. Alterations of the Gut Microbiome and TMAO Levels in Patients with Ulcerative Colitis. J. Clin. Med. 2024, 13, 5794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chaluvadi, M.R.; Reddy, R.; Motika, M.S.; Richardson, T.A.; Cashman, J.R.; Morgan, E.T. Hepatic Flavin-Containing Monooxygenase Gene Regulation in Different Mouse Inflammation Models. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Massey, W.; Mukherjee, P.; Nguyen, Q.T.; Mrdjen, M.; Wang, Z.; Brown, J.M.; Rieder, F. THE PATHOGENIC ROLE OF MICROBIAL TRIMETHYLAMINE IN IBD ASSOCIATED INTESTINAL FIBROSIS. Gastroenterology 2024, 166, S87–S88. [Google Scholar] [CrossRef]
- Kul, S.; Caliskan, Z.; Guvenc, T.S.; Celik, F.B.; Sarmis, A.; Atici, A.; Konal, O.; Akıl, M.; Cumen, A.S.; Bilgic, N.M.; et al. Gut Microbiota-Derived Metabolite Trimethylamine N-Oxide and Biomarkers of Inflammation Are Linked to Endothelial and Coronary Microvascular Function in Patients with Inflammatory Bowel Disease. Microvasc. Res. 2023, 146, 104458. [Google Scholar] [CrossRef] [PubMed]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross Sectional Evaluation of the Gut-Microbiome Metabolome Axis in an Italian Cohort of IBD Patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A. Alteration of the Gut Microbiome in Inflammatory Bowel Disease. Digestion 2023, 104, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, Z.; Xu, C.; Kan, S.; Chen, D. Disturbances of the Gut Microbiota and Microbiota-Derived Metabolites in Inflammatory Bowel Disease. Nutrients 2022, 14, 5140. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Fei, W.; Ye, Y.; Zhao, M.; Zheng, C. The Role of Short-Chain Fatty Acids in Immunity, Inflammation and Metabolism. Crit. Rev. Food Sci. Nutr. 2022, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Li, T.; Li, M.; Huang, S.; Qiu, Y.; Feng, R.; Zhang, S.; Chen, M.; Xiong, L.; Zeng, Z. Systematic Review and Meta-Analysis: Short-Chain Fatty Acid Characterization in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Han, S.; Kwon, J.; Ju, S.; Choi, T.; Kang, I.; Kim, S. Roles of Short-Chain Fatty Acids in Inflammatory Bowel Disease. Nutrients 2023, 15, 4466. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Y.; Zhao, X.; Shang, C.; Xiang, M.; Li, L.; Cui, X. Microbiota-Derived Short-Chain Fatty Acids: Implications for Cardiovascular and Metabolic Disease. Front. Cardiovasc. Med. 2022, 9, 900381. [Google Scholar] [CrossRef] [PubMed]
- Yukino-Iwashita, M.; Nagatomo, Y.; Kawai, A.; Taruoka, A.; Yumita, Y.; Kagami, K.; Yasuda, R.; Toya, T.; Ikegami, Y.; Masaki, N.; et al. Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure. J. Pers. Med. 2022, 12, 1805. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, R.; You, N.; Zhao, X.; Li, J.; Jiang, W. Butyric Acid Ameliorates Myocardial Fibrosis by Regulating M1/M2 Polarization of Macrophages and Promoting Recovery of Mitochondrial Function. Front. Nutr. 2022, 9, 875473. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, F.V.; Nielsen, H.; Mulvany, M.J.; Hessov, I. Short Chain Fatty Acids Dilate Isolated Human Colonic Resistance Arteries. Gut 1990, 31, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.J.; Larsen, A.M.; Homilius, C.; Gopalasingam, N.; Moeslund, N.; Berg-Hansen, K.; Boedtkjer, E.; Jensen, R.V.; Johannsen, M.; Hansen, J.; et al. Butyrate Increases Cardiac Output and Causes Vasorelaxation in a Healthy Porcine Model. Life Sci. 2025, 363, 123407. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, J.M.; Homilius, C.; Hansen, J.; Lassen, T.R.; Jespersen, N.R.; Jensen, R.V.; Boedtkjer, E.; Bøtker, H.E.; Nielsen, R. Short-Chain Fatty Acid Butyrate Is an Inotropic Agent With Vasorelaxant and Cardioprotective Properties. J. Am. Heart Assoc. 2024, 13, e033744. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, H.; Huo, P.; Shen, H.; Huang, Q.; Yang, L.; Liu, A.; Chen, G.; Tao, F.; Liu, K.; et al. The Association between Circulating Short-Chain Fatty Acids and Blood Pressure in Chinese Elderly Population. Sci. Rep. 2024, 14, 27062. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Eaton, D.M.; Berretta, R.M.; Liesinger, L.; Schittmayer, M.; Gindlhuber, J.; Wu, J.; Jeong, M.Y.; Lin, Y.H.; Borghetti, G.; et al. HDAC Inhibition Improves Cardiopulmonary Function in a Feline Model of Diastolic Dysfunction. Sci. Transl. Med. 2020, 12, eaay7205. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.Y.; Lin, Y.H.; Wennersten, S.A.; Demos-Davies, K.M.; Cavasin, M.A.; Mahaffey, J.H.; Monzani, V.; Saripalli, C.; Mascagni, P.; Reece, T.B.; et al. Histone Deacetylase Activity Governs Diastolic Dysfunction through a Nongenomic Mechanism. Sci. Transl. Med. 2018, 10, eaao0144. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Zeng, A.; Wu, I.; Greer-Short, A.; Farshidfar, F.; Budan, A.; Xu, E.; Shenwai, R.; Kozubov, M.; Li, C.; et al. Targeting HDAC6 to Treat Heart Failure with Preserved Ejection Fraction in Mice. Nat. Commun. 2024, 15, 1352. [Google Scholar] [CrossRef] [PubMed]
- Lkhagva, B.; Lin, Y.-K.; Kao, Y.-H.; Chazo, T.-F.; Chung, C.-C.; Chen, S.-A.; Chen, Y.-J. Novel Histone Deacetylase Inhibitor Modulates Cardiac Peroxisome Proliferator-Activated Receptors and Inflammatory Cytokines in Heart Failure. Pharmacology 2015, 96, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-H.; Liou, J.-P.; Chung, C.-C.; Lien, G.-S.; Kuo, C.-C.; Chen, S.-A.; Chen, Y.-J. Histone Deacetylase Inhibition Improved Cardiac Functions with Direct Antifibrotic Activity in Heart Failure. Int. J. Cardiol. 2013, 168, 4178–4183. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, Z.; Sun, Z.; Zhang, D.; Ma, X. Gut-Derived Short-Chain Fatty Acids Bridge Cardiac and Systemic Metabolism and Immunity in Heart Failure. J. Nutr. Biochem. 2023, 120, 109370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone Deacetylase (HDAC) Inhibition Improves Myocardial Function and Prevents Cardiac Remodeling in Diabetic Mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-Fat-Diet-Induced Obesity in Mice by Regulating G Protein-Coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.-J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARγ-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Sahuri-Arisoylu, M.; Brody, L.P.; Parkinson, J.R.; Parkes, H.; Navaratnam, N.; Miller, A.D.; Thomas, E.L.; Frost, G.; Bell, J.D. Reprogramming of Hepatic Fat Accumulation and “browning” of Adipose Tissue by the Short-Chain Fatty Acid Acetate. Int. J. Obes. 2016, 40, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Miles-Brown, J.; Pellizzon, M.; Ulman, E.; Ricci, M.; Zhang, L.; Patterson, A.D.; Vijay-Kumar, M.; Gewirtz, A.T. Lack of Soluble Fiber Drives Diet-Induced Adiposity in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G528–G541. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.S.P.; Obokata, M.; Rider, O.J. Obesity and Heart Failure with Preserved Ejection Fraction: New Insights and Pathophysiological Targets. Cardiovasc. Res. 2023, 118, 3434–3450. [Google Scholar] [CrossRef] [PubMed]
- Chulenbayeva, L.; Issilbayeva, A.; Sailybayeva, A.; Bekbossynova, M.; Kozhakhmetov, S.; Kushugulova, A. Short-Chain Fatty Acids and Their Metabolic Interactions in Heart Failure. Biomedicines 2025, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gu, Y.; Li, L.; Liu, T.; Song, X.; Sun, Y.; Cao, X.; Wang, B.; Jiang, K.; Cao, H. Bile Acid-Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside. Nutrients 2021, 13, 3143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lv, T.; Wang, X.; Wu, M.; Zhang, R.; Yang, X.; Fu, Y.; Liu, Z. Role of the Microbiota–Gut–Heart Axis between Bile Acids and Cardiovascular Disease. Biomed. Pharmacother. 2024, 174, 116567. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.H.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Wu, W.; Zhu, Y.; Liu, X. The Role of Bile Acids in Cardiovascular Diseases: From Mechanisms to Clinical Implications. Aging Dis. 2023, 14, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Tian, Z.; Mao, R.; Ma, R.; Luo, W.; Zhao, M.; Li, X.; Liu, Y.; Huang, K.; Xiang, L.; et al. Gut Microbiome-Generated Phenylacetylglutamine from Dietary Protein Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Model Possibly via Platelet Activation. J. Crohns Colitis 2023, 17, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.P.; Gogonea, V.; Sweet, W.; Mohan, M.L.; Singh, K.D.; Anderson, J.T.; Mallela, D.; Witherow, C.; Kar, N.; Stenson, K.; et al. Gut Microbe-Generated Phenylacetylglutamine Is an Endogenous Allosteric Modulator of Β2-Adrenergic Receptors. Nat. Commun. 2024, 15, 6696. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Nemet, I.; Li, X.S.; Wu, Y.; Haghikia, A.; Witkowski, M.; Koeth, R.A.; Demuth, I.; König, M.; Steinhagen-Thiessen, E.; et al. Prognostic Value of Gut Microbe-generated Metabolite Phenylacetylglutamine in Patients with Heart Failure. Eur. J. Heart Fail. 2024, 26, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Kong, B.; Zhu, J.; Huang, H.; Shuai, W. Phenylacetylglutamine Increases the Susceptibility of Ventricular Arrhythmias in Heart Failure Mice by Exacerbated Activation of the TLR4/AKT/mTOR Signaling Pathway. Int. Immunopharmacol. 2023, 116, 109795. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Li, D.; Shuai, W.; Kong, B.; Wang, X.; Tang, Y.; Huang, H.; Huang, C. Effects of Phenylacetylglutamine on the Susceptibility of Atrial Fibrillation in Overpressure-Induced HF Mice. Mol. Cell. Biol. 2024, 44, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.A.; Nemet, I.; Prasad Saha, P.; Haghikia, A.; Li, X.S.; Mohan, M.L.; Lovano, B.; Castel, L.; Witkowski, M.; Buffa, J.A.; et al. Gut Microbiota-Generated Phenylacetylglutamine and Heart Failure. Circ. Heart Fail. 2023, 16, e009972. [Google Scholar] [CrossRef] [PubMed]
- Verdugo-Meza, A.; Ye, J.; Dadlani, H.; Ghosh, S.; Gibson, D.L. Connecting the Dots Between Inflammatory Bowel Disease and Metabolic Syndrome: A Focus on Gut-Derived Metabolites. Nutrients 2020, 12, 1434. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Hernandez, M.M.; Vital, M.; Mohney, R.P.; Spector, T.D.; Valdes, A.M. Circulating Levels of the Anti-Oxidant Indoleproprionic Acid Are Associated with Higher Gut Microbiome Diversity. Gut Microbes 2019, 10, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, H.; Anjum, K.; Zhong, X.; Miao, S.; Zheng, G.; Liu, W.; Li, L. Dual Role of Indoles Derived from Intestinal Microbiota on Human Health. Front. Immunol. 2022, 13, 903526. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Béguin, E.P.; van den Eshof, B.L.; Hoogendijk, A.J.; Nota, B.; Mertens, K.; Meijer, A.B.; van den Biggelaar, M. Integrated Proteomic Analysis of Tumor Necrosis Factor α and Interleukin 1β-Induced Endothelial Inflammation. J. Proteom. 2019, 192, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving Functions of Endothelial Cells in Inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, K.; Guler, A.K.; Cakir, M.; Ozen, A.; Demirci, H.; Turker, T.; Demirbas, S.; Uygun, A.; Gulsen, M.; Bagci, S. Pulse Wave Velocity, Intima Media Thickness, and Flow-Mediated Dilatation in Patients with Normotensive Normoglycemic Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, Z.; Gokturk, H.S.; Caliskan, M.; Gullu, H.; Ciftci, O.; Ozgur, G.T.; Guven, A.; Selcuk, H. Impaired Coronary Microvascular and Left Ventricular Diastolic Function in Patients with Inflammatory Bowel Disease. Microvasc. Res. 2015, 97, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Rahman, H.; Webb, A.; Shah, A.M.; Perera, D. Untangling the Pathophysiologic Link between Coronary Microvascular Dysfunction and Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2021, 42, 4431–4441. [Google Scholar] [CrossRef] [PubMed]
- Cainzos-Achirica, M.; Glassner, K.; Zawahir, H.S.; Dey, A.K.; Agrawal, T.; Quigley, E.M.M.; Abraham, B.P.; Acquah, I.; Yahya, T.; Mehta, N.N.; et al. Inflammatory Bowel Disease and Atherosclerotic Cardiovascular Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Chen, G.; Cai, D.; Zhao, S.; Cheng, J.; Shen, H. Inflammatory Bowel Disease and Risk of Ischemic Heart Disease: An Updated Meta-Analysis of Cohort Studies. J. Am. Heart Assoc. 2017, 6, e005892. [Google Scholar] [CrossRef] [PubMed]
- Rungoe, C.; Basit, S.; Ranthe, M.F.; Wohlfahrt, J.; Langholz, E.; Jess, T. Risk of Ischaemic Heart Disease in Patients with Inflammatory Bowel Disease: A Nationwide Danish Cohort Study. Gut 2013, 62, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I. Mechanisms Linking Red Blood Cell Disorders and Cardiovascular Diseases. BioMed Res. Int. 2015, 2015, 682054. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-N.; Yao, Y.; Ju, S.-Y. Heart Rate Variability and Inflammatory Bowel Disease in Humans: A Systematic Review and Meta-Analysis. Medicine 2020, 99, e23430. [Google Scholar] [CrossRef] [PubMed]
- Aghdasi-Bornaun, H.; Kutluk, G.; Keskindemirci, G.; Öztarhan, K.; Dedeoğlu, R.; Yılmaz, N.; Tosun, Ö. Evaluation of Autonomic Nervous System Functions in Frame of Heart Rate Variability in Children with Inflammatory Bowel Disease in Remission. Turk. J. Pediatr. 2018, 60, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, S.; Dantzer, C.; Mondillon, L.; Trocme, C.; Gauchez, A.-S.; Ducros, V.; Mathieu, N.; Toussaint, B.; Fournier, A.; Canini, F.; et al. Relationship between Vagal Tone, Cortisol, TNF-Alpha, Epinephrine and Negative Affects in Crohn’s Disease and Irritable Bowel Syndrome. PLoS ONE 2014, 9, e105328. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zhou, K.; Wang, S.; Li, P.; Chen, S.; Lin, G.; Zhao, Y.; Wang, T. Involvement of MAPK/NF-κB Signaling in the Activation of the Cholinergic Anti-Inflammatory Pathway in Experimental Colitis by Chronic Vagus Nerve Stimulation. PLoS ONE 2013, 8, e69424. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, W.J.; Ulloa, L. The Alpha7 Nicotinic Acetylcholine Receptor as a Pharmacological Target for Inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.E.; Holmes, G.M.; Rogers, R.C. TNF(Alpha) Modulation of Visceral and Spinal Sensory Processing. Curr. Pharm. Des. 2005, 11, 1391–1409. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.E.; Rogers, R.C. TNFalpha: A Trigger of Autonomic Dysfunction. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2008, 14, 53–67. [Google Scholar] [CrossRef]
- Florea, V.G.; Cohn, J.N. The Autonomic Nervous System and Heart Failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure: Pathophysiology and Therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, M.; Liu, Y.; Ge, C.; Lu, Y.; Shen, H.; Zhu, L. Prevalence of Metabolic Syndrome in Patients with Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. BMJ Open 2024, 14, e074659. [Google Scholar] [CrossRef] [PubMed]
- Del Pinto, R.; Pietropaoli, D.; Chandar, A.K.; Ferri, C.; Cominelli, F. Association Between Inflammatory Bowel Disease and Vitamin D Deficiency: A Systematic Review and Meta-Analysis. Inflamm. Bowel Dis. 2015, 21, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Bouguen, G.; Laharie, D.; Pellet, G.; Savoye, G.; Gilletta, C.; Michiels, C.; Buisson, A.; Fumery, M.; Trochu, J.-N.; et al. Iron Deficiency in Patients with Inflammatory Bowel Diseases: A Prospective Multicenter Cross-Sectional Study. Dig. Dis. Sci. 2022, 67, 5637–5646. [Google Scholar] [CrossRef] [PubMed]
- Bezzio, C.; Brinch, D.; Ribaldone, D.G.; Cappello, M.; Ruzzon, N.; Vernero, M.; Scalvini, D.; Loy, L.; Donghi, S.; Ciminnisi, S.; et al. Prevalence, Risk Factors and Association with Clinical Outcomes of Malnutrition and Sarcopenia in Inflammatory Bowel Disease: A Prospective Study. Nutrients 2024, 16, 3983. [Google Scholar] [CrossRef] [PubMed]
- Fatani, H.; Olaru, A.; Stevenson, R.; Alharazi, W.; Jafer, A.; Atherton, P.; Brook, M.; Moran, G. Systematic Review of Sarcopenia in Inflammatory Bowel Disease. Clin. Nutr. 2023, 42, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Nakamura, S.; Miyazaki, T.; Kakimoto, K.; Fukunishi, S.; Asai, A.; Nishiguchi, S.; Higuchi, K. Inflammatory Bowel Disease and Sarcopenia: Its Mechanism and Clinical Importance. J. Clin. Med. 2021, 10, 4214. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.M.; Grimes, J.B.; Alonso, D.; Alonso, M.; Gay, W.A. Adverse Hemodynamic and Ultrastructural Changes in Dog Hearts Subjected to Protein-Calorie Malnutrition. Am. Heart J. 1979, 97, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, D.; Theodorakis, N.; Hitas, C.; Kreouzi, M.; Pantos, I.; Vamvakou, G.; Nikolaou, M. Sarcopenia and Cardiogeriatrics: The Links Between Skeletal Muscle Decline and Cardiovascular Aging. Nutrients 2025, 17, 282. [Google Scholar] [CrossRef] [PubMed]
- Damluji, A.A.; Alfaraidhy, M.; AlHajri, N.; Rohant, N.N.; Kumar, M.; Al Malouf, C.; Bahrainy, S.; Ji Kwak, M.; Batchelor, W.B.; Forman, D.E.; et al. Sarcopenia and Cardiovascular Diseases. Circulation 2023, 147, 1534–1553. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xu, J.; Wang, Y.; Jiang, B.; Xu, X.; Lan, Y.; Wang, J.; Lin, X. Prevalence of Sarcopenia and Its Association with Clinical Outcomes in Heart Failure: An Updated Meta-Analysis and Systematic Review. Clin. Cardiol. 2023, 46, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Adejumo, A.C.; Adejumo, K.L.; Adegbala, O.M.; Chinedozi, I.; Ndansi, J.; Akanbi, O.; Onyeakusi, N.E.; Ogundipe, O.A.; Bob-Manuel, T.; Adeboye, A. Protein-Energy Malnutrition and Outcomes of Hospitalizations for Heart Failure in the USA. Am. J. Cardiol. 2019, 123, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Mahadea, D.; Adamczewska, E.; Ratajczak, A.E.; Rychter, A.M.; Zawada, A.; Eder, P.; Dobrowolska, A.; Krela-Kaźmierczak, I. Iron Deficiency Anemia in Inflammatory Bowel Diseases-A Narrative Review. Nutrients 2021, 13, 4008. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, S.; Kalogeropoulos, A.P. Markers of Iron Metabolism and Outcomes in Patients with Heart Failure: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 5645. [Google Scholar] [CrossRef] [PubMed]
- Manceau, H.; Ausseil, J.; Masson, D.; Feugeas, J.-P.; Sablonniere, B.; Guieu, R.; Puy, H.; Peoc’h, K. Neglected Comorbidity of Chronic Heart Failure: Iron Deficiency. Nutrients 2022, 14, 3214. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; von Haehling, S.; Butler, J.; Cleland, J.G.F.; Ponikowski, P.; Anker, S.D. Iron Deficiency and Cardiovascular Disease. Eur. Heart J. 2023, 44, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron Deficiency Impairs Contractility of Human Cardiomyocytes through Decreased Mitochondrial Function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Luo, A.; Park, K.C.; Loonat, A.A.; Lakhal-Littleton, S.; Robbins, P.A.; Swietach, P. Iron-Deficiency Anemia Reduces Cardiac Contraction by Downregulating RyR2 Channels and Suppressing SERCA Pump Activity. JCI Insight 2019, 4, e125618. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Moss, A.C. Vitamin D in Inflammatory Bowel Disease: More than Just a Supplement. Curr. Opin. Gastroenterol. 2018, 34, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.; Cooper, S.C.; Ghosh, S.; Hewison, M. The Role of Vitamin D in Inflammatory Bowel Disease: Mechanism to Management. Nutrients 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Latic, N.; Erben, R.G. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Marginean, O. Links between Vitamin D Deficiency and Cardiovascular Diseases. BioMed Res. Int. 2015, 2015, 109275. [Google Scholar] [CrossRef] [PubMed]
- Maryam; Varghese, T.P.; B, T. Unraveling the Complex Pathophysiology of Heart Failure: Insights into the Role of Renin-Angiotensin-Aldosterone System (RAAS) and Sympathetic Nervous System (SNS). Curr. Probl. Cardiol. 2024, 49, 102411. [Google Scholar] [CrossRef] [PubMed]
- Wöbke, T.K.; Sorg, B.L.; Steinhilber, D. Vitamin D in Inflammatory Diseases. Front. Physiol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Busa, V.; Dardeir, A.; Marudhai, S.; Patel, M.; Valaiyaduppu Subas, S.; Ghani, M.R.; Cancarevic, I. Role of Vitamin D Supplementation in Heart Failure Patients with Vitamin D Deficiency and Its Effects on Clinical Outcomes: A Literature Review. Cureus 2020, 12, e10840. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.B.; Jones, H.W.; Gordon, A.C. Selenium Deficiency, Reversible Cardiomyopathy and Short-Term Intravenous Feeding. Postgrad. Med. J. 1994, 70, 235–236. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Yamaguchi, T.; Tanaka, H.; Hashimoto, S.; Yang, M.; Takahashi, T. TPN-Induced Fulminant Beriberi: A Report on Our Experience and a Review of the Literature. Surg. Today 1996, 26, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Jin, T.; Yi, C.; Ocansey, D.K.W.; Mao, F. The Role of NOD2 in Intestinal Immune Response and Microbiota Modulation: A Therapeutic Target in Inflammatory Bowel Disease. Int. Immunopharmacol. 2022, 113, 109466. [Google Scholar] [CrossRef] [PubMed]
- Masaki, S.; Masuta, Y.; Honjo, H.; Kudo, M.; Watanabe, T. NOD2-Mediated Dual Negative Regulation of Inflammatory Responses Triggered by TLRs in the Gastrointestinal Tract. Front. Immunol. 2024, 15, 1433620. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Salim, M.; Zhou, H.; Bian, Z.; Dai, J.; Yuan, Y.; Deng, W.; Zhang, J.; Zhang, R.; Wu, Q.; et al. NOD2 Deletion Promotes Cardiac Hypertrophy and Fibrosis Induced by Pressure Overload. Lab. Investig. 2013, 93, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Li, J.; Gong, L.; Shao, S.; Chen, H.; He, P.; Yan, J. Casual Effect of Ulcerative Colitis on Chronic Heart Failure: Results from a Bidirectional Mendelian Randomization Study. BMC Gastroenterol. 2025, 25, 95. [Google Scholar] [CrossRef] [PubMed]
- Minea, H.; Singeap, A.-M.; Minea, M.; Juncu, S.; Muzica, C.; Sfarti, C.V.; Girleanu, I.; Chiriac, S.; Miftode, I.D.; Stanciu, C.; et al. The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2024, 25, 8420. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hu, X.; Guo, X.; Zhang, B.; Xu, W.; Jiang, H. Promoting Effects of IL-23 on Myocardial Ischemia and Reperfusion Are Associated with Increased Expression of IL-17A and Upregulation of the JAK2-STAT3 Signaling Pathway. Mol. Med. Rep. 2017, 16, 9309–9316. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Bahls, M.; Könemann, S.; Markus, M.R.P.; Wenzel, K.; Friedrich, N.; Nauck, M.; Völzke, H.; Steveling, A.; Janowitz, D.; Grabe, H.-J.; et al. Brain-Derived Neurotrophic Factor Is Related with Adverse Cardiac Remodeling and High NTproBNP. Sci. Rep. 2019, 9, 15421. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New Players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Narumi, T.; Watanabe, T.; Otaki, Y.; Takahashi, T.; Aono, T.; Goto, J.; Toshima, T.; Sugai, T.; Wanezaki, M.; et al. The Association between microRNA-21 and Hypertension-Induced Cardiac Remodeling. PLoS ONE 2020, 15, e0226053. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 Contributes to Myocardial Disease by Stimulating MAP Kinase Signalling in Fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models. Cells 2021, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MiR-155 Knockout in Fibroblasts Improves Cardiac Remodeling by Targeting Tumor Protein P53-Inducible Nuclear Protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; Van Leeuwen, R.E.W.; Custers, K.; Peters, T.; Hazebroek, M.; Stöger, L.; Wijnands, E.; et al. Macrophage MicroRNA-155 Promotes Cardiac Hypertrophy and Failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Albar, Z.; Albakri, M.; Hajjari, J.; Karnib, M.; Janus, S.E.; Al-Kindi, S.G. Inflammatory Markers and Risk of Heart Failure with Reduced to Preserved Ejection Fraction. Am. J. Cardiol. 2022, 167, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Durham, H.; Glover, L.; Ather, O.; Phillips, V.; Nemes, S.; Cousens, L.; Blomgran, P.; Ambery, P. Metabolic Adverse Events Associated with Systemic Corticosteroid Therapy-a Systematic Review and Meta-Analysis. BMJ Open 2022, 12, e061476. [Google Scholar] [CrossRef] [PubMed]
- Pujades-Rodriguez, M.; Morgan, A.W.; Cubbon, R.M.; Wu, J. Dose-Dependent Oral Glucocorticoid Cardiovascular Risks in People with Immune-Mediated Inflammatory Diseases: A Population-Based Cohort Study. PLoS Med. 2020, 17, e1003432. [Google Scholar] [CrossRef] [PubMed]
- Kirchgesner, J.; Nyboe Andersen, N.; Carrat, F.; Jess, T.; Beaugerie, L.; BERENICE Study Group. Risk of Acute Arterial Events Associated with Treatment of Inflammatory Bowel Diseases: Nationwide French Cohort Study. Gut 2020, 69, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L.; O’Bryant, C.L.; Cheng, D.; Dow, T.J.; Ky, B.; Stein, C.M.; Spencer, A.P.; Trupp, R.J.; Lindenfeld, J. Drugs That May Cause or Exacerbate Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2016, 134, e32–e69. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shokravi, A.; Luo, Y.; Rabkin, S.W. Cellular and Molecular Mechanisms Explaining the Link Between Inflammatory Bowel Disease and Heart Failure. Cells 2025, 14, 1124. https://doi.org/10.3390/cells14141124
Shokravi A, Luo Y, Rabkin SW. Cellular and Molecular Mechanisms Explaining the Link Between Inflammatory Bowel Disease and Heart Failure. Cells. 2025; 14(14):1124. https://doi.org/10.3390/cells14141124
Chicago/Turabian StyleShokravi, Arveen, Yuchen Luo, and Simon W. Rabkin. 2025. "Cellular and Molecular Mechanisms Explaining the Link Between Inflammatory Bowel Disease and Heart Failure" Cells 14, no. 14: 1124. https://doi.org/10.3390/cells14141124
APA StyleShokravi, A., Luo, Y., & Rabkin, S. W. (2025). Cellular and Molecular Mechanisms Explaining the Link Between Inflammatory Bowel Disease and Heart Failure. Cells, 14(14), 1124. https://doi.org/10.3390/cells14141124