
Placental Inflammation in Preterm Premature Rupture of Membranes and Risk of Neurodevelopmental Disorders



Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. pPROM Etiology and Risk Factors
2.1. Ascending Infections
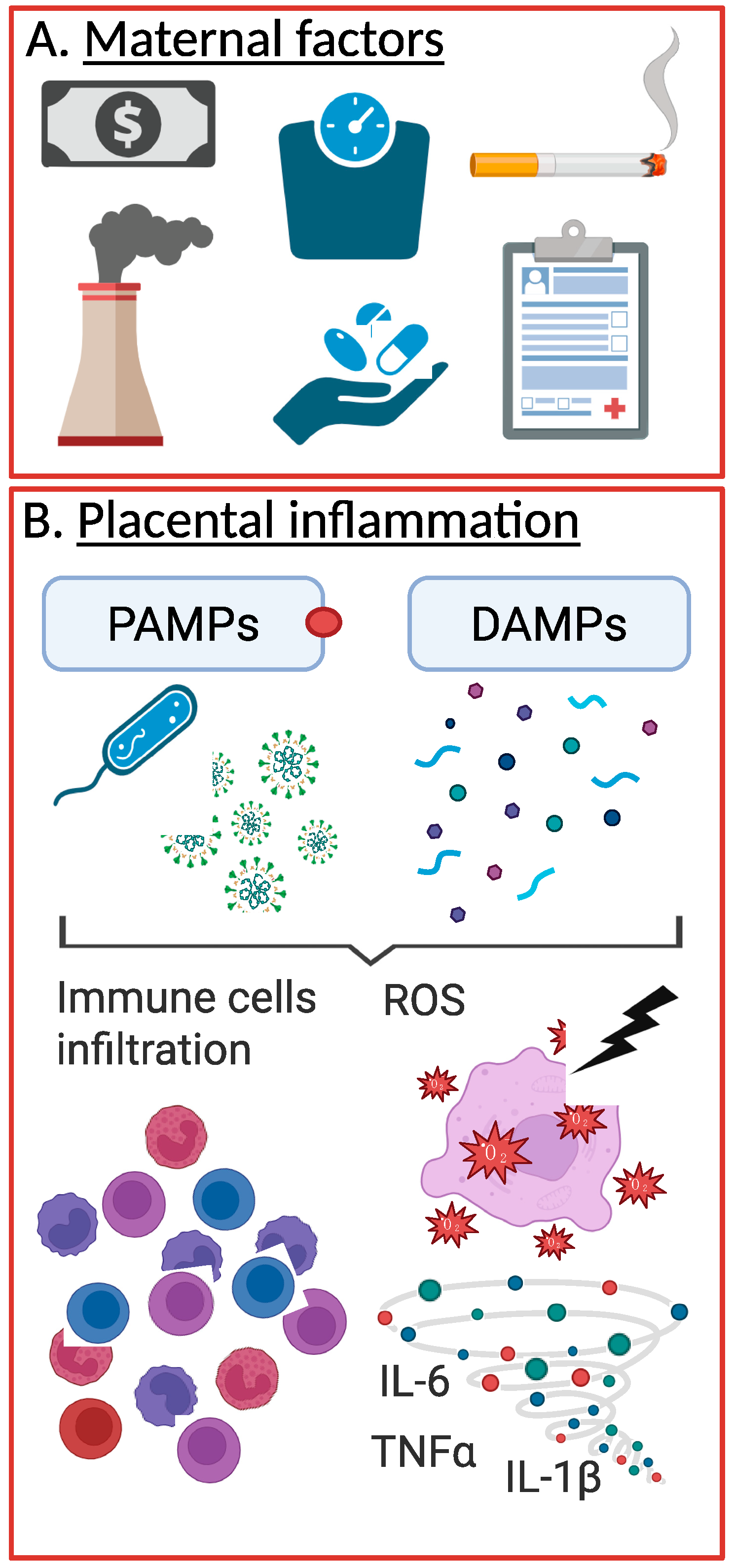
2.2. Causes of Placental Inflammation
2.2.1. Pathogen-Associated Inflammation
2.2.2. Sterile Inflammation
2.2.3. Pathogen-Driven and Sterile Inflammation in pPROM: Acute and Chronic Inflammatory Response
3. Placental Inflammation and Neurodevelopmental Disruptions
3.1. Animal Models of Prenatal Inflammation’s Impact on Neurodevelopment
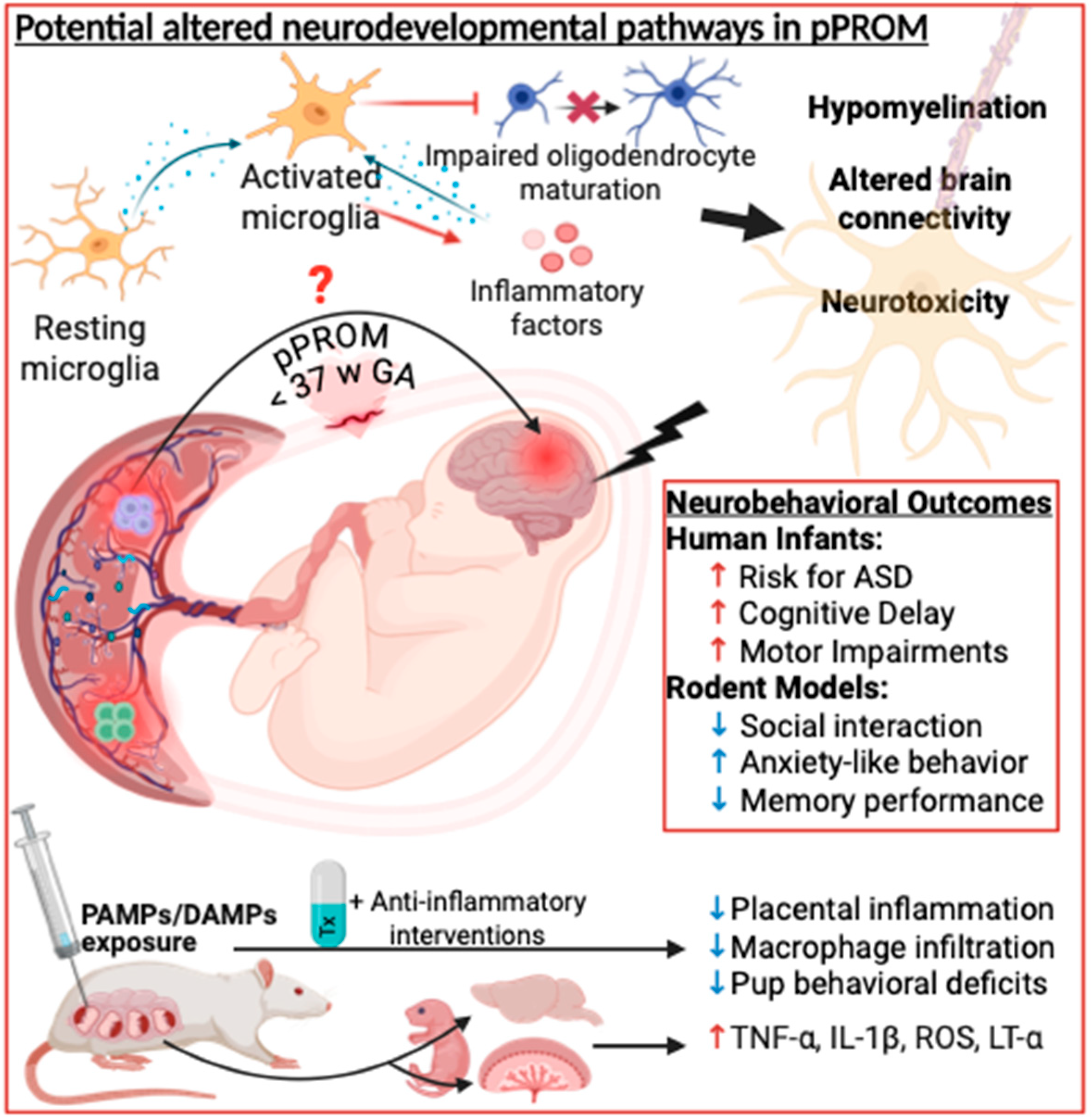
Summary and Future Directions in Animal Models
3.2. Human Studies
4. Perspectives
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weng, J.; Couture, C.; Girard, S. Innate and Adaptive Immune Systems in Physiological and Pathological Pregnancy. Biology 2023, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Morniroli, D.; Tiraferri, V.; Maiocco, G.; De Rose, D.U.; Cresi, F.; Coscia, A.; Mosca, F.; Giannì, M.L. Beyond survival: The lasting effects of premature birth. Front. Pediatr. 2023, 11, 1213243. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Jenkins, S.M.; Hong, P.L. Preterm and Term Prelabor Rupture of Membranes (PPROM and PROM). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Manuck, T.A.; Varner, M.W. Neonatal and early childhood outcomes following early vs later preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2014, 211, 308.e1–308.e6. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.L.; Thompson, P.M. Typical and atypical brain development: A review of neuroimaging studies. Dialogues Clin. Neurosci. 2013, 15, 359–384. [Google Scholar] [CrossRef]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Han, T.H.; Chae, K.Y.; Han, B.; Kim, J.H.; Ha, E.K.; Rhie, S.; Han, M.Y. Early onset and increasing disparities in neurodevelopmental delays from birth to age 6 in children from low socioeconomic backgrounds. J. Neurodev. Disord. 2024, 16, 60. [Google Scholar] [CrossRef]
- Milner, K.M.; Duke, T.; Steer, A.C.; Kado, J.H.; Koyamaibole, L.; Kaarira, R.; Namudu, K.; Woolfenden, S.; Miller, A.E.; O’Heir, K.E.; et al. Neurodevelopmental outcomes for high-risk neonates in a low-resource setting. Arch. Dis. Child. 2017, 102, 1063–1069. [Google Scholar] [CrossRef]
- Murillo, C.; Eixarch, E.; Rueda, C.; Larroya, M.; Boada, D.; Grau, L.; Ponce, J.; Aldecoa, V.; Monterde, E.; Ferrero, S.; et al. Evidence of brain injury in fetuses of mothers with preterm labor with intact membranes and preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2025, 232, 114.e111–114.e124. [Google Scholar] [CrossRef]
- Ginsberg, Y.; Khatib, N.; Weiner, Z.; Beloosesky, R. Maternal Inflammation, Fetal Brain Implications and Suggested Neuroprotection: A Summary of 10 Years of Research in Animal Models. Rambam. Maimonides Med. J. 2017, 8, e0028. [Google Scholar] [CrossRef]
- Kwon, H.K.; Choi, G.B.; Huh, J.R. Maternal inflammation and its ramifications on fetal neurodevelopment. Trends Immunol. 2022, 43, 230–244. [Google Scholar] [CrossRef]
- Garfinkle, J.; Miller, S.P. The Placenta and Neurodevelopment in Preterm Newborns. NeoReviews 2018, 19, e456–e466. [Google Scholar] [CrossRef]
- Venkatesh, K.K.; Leviton, A.; Hecht, J.L.; Joseph, R.M.; Douglass, L.M.; Frazier, J.A.; Daniels, J.L.; Fry, R.C.; O’Shea, T.M.; Kuban, K.C.K. Histologic chorioamnionitis and risk of neurodevelopmental impairment at age 10 years among extremely preterm infants born before 28 weeks of gestation. Am. J. Obstet. Gynecol. 2020, 223, 745.e1–745.e10. [Google Scholar] [CrossRef]
- Liu, K.; Huang, Y.; Zhu, Y.; Zhao, Y.; Kong, X. The role of maternal immune activation in immunological and neurological pathogenesis of autism. J. Neurorestoratology 2023, 11, 100030. [Google Scholar] [CrossRef]
- Menon, R.; Behnia, F.; Polettini, J.; Richardson, L.S. Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes. Semin. Immunopathol. 2020, 42, 431–450. [Google Scholar] [CrossRef]
- Menon, R.; Richardson, L.S.; Lappas, M. Fetal membrane architecture, aging and inflammation in pregnancy and parturition. Placenta 2019, 79, 40–45. [Google Scholar] [CrossRef]
- Ward, E.J.; Bert, S.; Fanti, S.; Malone, K.M.; Maughan, R.T.; Gkantsinikoudi, C.; Prin, F.; Volpato, L.K.; Piovezan, A.P.; Graham, G.J.; et al. Placental Inflammation Leads to Abnormal Embryonic Heart Development. Circulation 2023, 147, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Heerema-McKenney, A. Defense and infection of the human placenta. APMIS 2018, 126, 570–588. [Google Scholar] [CrossRef]
- Cornish, E.F.; McDonnell, T.; Williams, D.J. Chronic Inflammatory Placental Disorders Associated With Recurrent Adverse Pregnancy Outcome. Front. Immunol. 2022, 13, 825075. [Google Scholar] [CrossRef] [PubMed]
- Salafia, C.; Popek, E. Inflammatory and Vascular Placental Pathology. Glob. Libr. Women’s Med. 2008. [Google Scholar] [CrossRef]
- Bouvier, D.; Forest, J.C.; Blanchon, L.; Bujold, E.; Pereira, B.; Bernard, N.; Gallot, D.; Sapin, V.; Giguère, Y. Risk Factors and Outcomes of Preterm Premature Rupture of Membranes in a Cohort of 6968 Pregnant Women Prospectively Recruited. J. Clin. Med. 2019, 8, 1987. [Google Scholar] [CrossRef]
- Vidal, M.S., Jr.; Lintao, R.C.V.; Severino, M.E.L.; Tantengco, O.A.G.; Menon, R. Spontaneous preterm birth: Involvement of multiple feto-maternal tissues and organ systems, differing mechanisms, and pathways. Front. Endocrinol. 2022, 13, 1015622. [Google Scholar] [CrossRef]
- Shen, T.T.; DeFranco, E.A.; Stamilio, D.M.; Chang, J.J.; Muglia, L.J. A population-based study of race-specific risk for preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2008, 199, 373.e371–373.e377. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; Gallagher, K.; Beck, C.; Kumar, R.; Gernand, A.D. Maternal-Fetal Inflammation in the Placenta and the Developmental Origins of Health and Disease. Front. Immunol. 2020, 11, 531543. [Google Scholar] [CrossRef]
- Humberg, A.; Fortmann, I.; Siller, B.; Kopp, M.V.; Herting, E.; Göpel, W.; Härtel, C. Preterm birth and sustained inflammation: Consequences for the neonate. Semin. Immunopathol. 2020, 42, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Bennet, L.; Dhillon, S.; Lear, C.A.; van den Heuij, L.; King, V.; Dean, J.M.; Wassink, G.; Davidson, J.O.; Gunn, A.J. Chronic inflammation and impaired development of the preterm brain. J. Reprod. Immunol. 2018, 125, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.J.; Brochu, M.E.; Bergeron, J.D.; Segura, M.; Sébire, G. Causal role of group B Streptococcus-induced acute chorioamnionitis in intrauterine growth retardation and cerebral palsy-like impairments. J. Dev. Orig. Health Dis. 2019, 10, 595–602. [Google Scholar] [CrossRef]
- Oh, K.J.; Lee, K.A.; Sohn, Y.K.; Park, C.W.; Hong, J.S.; Romero, R.; Yoon, B.H. Intraamniotic infection with genital mycoplasmas exhibits a more intense inflammatory response than intraamniotic infection with other microorganisms in patients with preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2010, 203, 211.e211–211.e218. [Google Scholar] [CrossRef]
- Viscardi, R.M. Ureaplasma species: Role in diseases of prematurity. Clin. Perinatol. 2010, 37, 393–409. [Google Scholar] [CrossRef]
- Elovitz, M.A.; Gajer, P.; Riis, V.; Brown, A.G.; Humphrys, M.S.; Holm, J.B.; Ravel, J. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat. Commun. 2019, 10, 1305. [Google Scholar] [CrossRef]
- Gudnadottir, U.; Debelius, J.W.; Du, J.; Hugerth, L.W.; Danielsson, H.; Schuppe-Koistinen, I.; Fransson, E.; Brusselaers, N. The vaginal microbiome and the risk of preterm birth: A systematic review and network meta-analysis. Sci. Rep. 2022, 12, 7926. [Google Scholar] [CrossRef]
- Daskalakis, G.; Psarris, A.; Koutras, A.; Fasoulakis, Z.; Prokopakis, I.; Varthaliti, A.; Karasmani, C.; Ntounis, T.; Domali, E.; Theodora, M.; et al. Maternal Infection and Preterm Birth: From Molecular Basis to Clinical Implications. Children 2023, 10, 907. [Google Scholar] [CrossRef] [PubMed]
- Hoo, R.; Nakimuli, A.; Vento-Tormo, R. Innate Immune Mechanisms to Protect Against Infection at the Human Decidual-Placental Interface. Front. Immunol. 2020, 11, 2070. [Google Scholar] [CrossRef] [PubMed]
- Semmes, E.C.; Coyne, C.B. Innate immune defenses at the maternal-fetal interface. Curr. Opin. Immunol. 2022, 74, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Holguín, V.J.; González-García, L.D.; Velázquez-Cervantes, M.A.; Arévalo-Romero, H.; De Jesús-González, L.A.; Helguera-Repetto, A.C.; León-Reyes, G.; Salazar, M.I.; Cedillo-Barrón, L.; León-Juárez, M. Collateral Damage in the Placenta during Viral Infection in Pregnancy: A Possible Mechanism for Vertical Transmission and an Adverse Pregnancy Outcome. Diseases 2024, 12, 59. [Google Scholar] [CrossRef]
- Abrahams, V.M.; Mor, G. Toll-Like Receptors and Pregnancy. In Immunology of Pregnancy; Mor, G., Ed.; Springer: New York, NY, USA, 2006; pp. 15–25. [Google Scholar]
- Nadeau-Vallée, M.; Obari, D.; Palacios, J.; Brien, M.; Duval, C.; Chemtob, S.; Girard, S. Sterile inflammation and pregnancy complications: A review. Reproduction 2016, 152, R277–R292. [Google Scholar] [CrossRef]
- Brien, M.E.; Baker, B.; Duval, C.; Gaudreault, V.; Jones, R.L.; Girard, S. Alarmins at the maternal-fetal interface: Involvement of inflammation in placental dysfunction and pregnancy complications. Can. J. Physiol. Pharmacol. 2019, 97, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.C.; Heazell, A.E.P.; Sibley, C.; Wright, R.; Bischof, H.; Beards, F.; Guevara, T.; Girard, S.; Jones, R.L. Hypoxia and oxidative stress induce sterile placental inflammation in vitro. Sci. Rep. 2021, 11, 7281. [Google Scholar] [CrossRef]
- Woods, J.R., Jr. Reactive oxygen species and preterm premature rupture of membranes-a review. Placenta 2001, 22 (Suppl. A), S38–S44. [Google Scholar] [CrossRef]
- Dixon, C.L.; Richardson, L.; Sheller-Miller, S.; Saade, G.; Menon, R. A distinct mechanism of senescence activation in amnion epithelial cells by infection, inflammation, and oxidative stress. Am. J. Reprod. Immunol. 2018, 79, 29193446. [Google Scholar] [CrossRef]
- Gal, H.; Lysenko, M.; Stroganov, S.; Vadai, E.; Youssef, S.A.; Tzadikevitch-Geffen, K.; Rotkopf, R.; Biron-Shental, T.; de Bruin, A.; Neeman, M.; et al. Molecular pathways of senescence regulate placental structure and function. Embo J. 2019, 38, 29193446. [Google Scholar] [CrossRef]
- Kajdy, A.; Modzelewski, J.; Cymbaluk-Płoska, A.; Kwiatkowska, E.; Bednarek-Jędrzejek, M.; Borowski, D.; Stefańska, K.; Rabijewski, M.; Torbé, A.; Kwiatkowski, S. Molecular Pathways of Cellular Senescence and Placental Aging in Late Fetal Growth Restriction and Stillbirth. Int. J. Mol. Sci. 2021, 22, 4186. [Google Scholar] [CrossRef]
- Menon, R. Oxidative stress damage as a detrimental factor in preterm birth pathology. Front. Immunol. 2014, 5, 567. [Google Scholar] [CrossRef] [PubMed]
- Brien, M.E.; Duval, C.; Palacios, J.; Boufaied, I.; Hudon-Thibeault, A.A.; Nadeau-Vallée, M.; Vaillancourt, C.; Sibley, C.P.; Abrahams, V.M.; Jones, R.L.; et al. Uric Acid Crystals Induce Placental Inflammation and Alter Trophoblast Function via an IL-1-Dependent Pathway: Implications for Fetal Growth Restriction. J. Immunol. 2017, 198, 443–451. [Google Scholar] [CrossRef]
- Brien, M.E.; Hughes, K.; Girard, S. Prenatal administration of IL-1Ra attenuate the neurodevelopmental impacts following non-pathogenic inflammation during pregnancy. Sci. Rep. 2021, 11, 23404. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.A.; Marteinsdottir, I.; Josefsson, A.; Sydsjö, G.; Theodorsson, E.; Rodriguez-Martinez, H. Epigenetic modifications appear in the human placenta following anxiety and depression during pregnancy. Placenta 2023, 140, 72–79. [Google Scholar] [CrossRef]
- Vornic, I.; Buciu, V.; Furau, C.G.; Gaje, P.N.; Ceausu, R.A.; Dumitru, C.S.; Barb, A.C.; Novacescu, D.; Cumpanas, A.A.; Latcu, S.C.; et al. Oxidative Stress and Placental Pathogenesis: A Contemporary Overview of Potential Biomarkers and Emerging Therapeutics. Int. J. Mol. Sci. 2024, 25, 12195. [Google Scholar] [CrossRef] [PubMed]
- Tita, A.T.; Andrews, W.W. Diagnosis and management of clinical chorioamnionitis. Clin. Perinatol. 2010, 37, 339–354. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Ancel, P.Y.; Foix-L’Hélias, L.; Marret, S.; Kayem, G. Impact of clinical and/or histological chorioamnionitis on neurodevelopmental outcomes in preterm infants: A literature review. J. Gynecol. Obstet. Hum. Reprod. 2017, 46, 307–316. [Google Scholar] [CrossRef]
- Orsaria, M.; Liviero, S.; Rossetti, E.; Pittini, C.; Driul, L.; Londero, A.P.; Mariuzzi, L. Placental acute inflammation infiltrates and pregnancy outcomes: A retrospective cohort study. Sci. Rep. 2021, 11, 24165. [Google Scholar] [CrossRef]
- Enninga, E.A.L.; Raber, P.; Quinton, R.A.; Ruano, R.; Ikumi, N.; Gray, C.M.; Johnson, E.L.; Chakraborty, R.; Kerr, S.E. Maternal T Cells in the Human Placental Villi Support an Allograft Response during Noninfectious Villitis. J. Immunol. 2020, 204, 2931–2939. [Google Scholar] [CrossRef]
- Zhou, J.; Tong, J.; Ru, X.; Teng, Y.; Geng, M.; Yan, S.; Tao, F.; Huang, K. Placental inflammatory cytokines mRNA expression and preschool children’s cognitive performance: A birth cohort study in China. BMC Med. 2023, 21, 449. [Google Scholar] [CrossRef]
- Menon, R.; Richardson, L.S. Preterm prelabor rupture of the membranes: A disease of the fetal membranes. Semin. Perinatol. 2017, 41, 409–419. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Block, C.L.; Bolton, J.L.; Hanamsagar, R.; Tran, P.K. Beyond infection—Maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp. Neurol. 2018, 299, 241–251. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol. 2012, 33, 267–286. [Google Scholar] [CrossRef]
- Girard, S.; Sebire, G. Transplacental Transfer of Interleukin-1 Receptor Agonist and Antagonist Following Maternal Immune Activation. Am. J. Reprod. Immunol. 2016, 75, 8–12. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef] [PubMed]
- da Silva, C.S.; Calió, M.L.; Mosini, A.C.; Pires, J.M.; Rêgo, D.; Mello, L.E.; Leslie, A. LPS-Induced Systemic Neonatal Inflammation: Blockage of P2X7R by BBG Decreases Mortality on Rat Pups and Oxidative Stress in Hippocampus of Adult Rats. Front. Behav. Neurosci. 2019, 13, 240. [Google Scholar] [CrossRef]
- Bell, M.J.; Hallenbeck, J.M. Effects of intrauterine inflammation on developing rat brain. J. Neurosci. Res. 2002, 70, 570–579. [Google Scholar] [CrossRef]
- Huleihel, M.; Golan, H.; Hallak, M. Intrauterine infection/inflammation during pregnancy and offspring brain damages: Possible mechanisms involved. Reprod. Biol. Endocrinol. 2004, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Aria, F.; Bonini, S.A.; Cattaneo, V.; Premoli, M.; Mastinu, A.; Maccarinelli, G.; Memo, M. Brain Structural and Functional Alterations in Mice Prenatally Exposed to LPS Are Only Partially Rescued by Anti-Inflammatory Treatment. Brain Sci. 2020, 10, 620. [Google Scholar] [CrossRef]
- Girard, S.; Kadhim, H.; Beaudet, N.; Sarret, P.; Sébire, G. Developmental motor deficits induced by combined fetal exposure to lipopolysaccharide and early neonatal hypoxia/ischemia: A novel animal model for cerebral palsy in very premature infants. Neuroscience 2009, 158, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qi, F.; Song, D.; He, Z.; Zuo, Z.; Yang, Y.; Liu, Q.; Hu, S.; Wang, X.; Zheng, X.; et al. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J. Neuroinflamm. 2018, 15, 228. [Google Scholar] [CrossRef] [PubMed]
- Giovanoli, S.; Notter, T.; Richetto, J.; Labouesse, M.A.; Vuillermot, S.; Riva, M.A.; Meyer, U. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflamm. 2015, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.K.; Ince, L.M.; Gullapalli, S.; Fonken, L.K. Neuroimmune and behavioral changes elicited by maternal immune activation in mice are ameliorated by early postnatal immune stimulation. Brain Behav. Immun. 2025, 127, 375–386. [Google Scholar] [CrossRef]
- Arsenault, D.; St-Amour, I.; Cisbani, G.; Rousseau, L.S.; Cicchetti, F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav. Immun. 2014, 38, 77–90. [Google Scholar] [CrossRef]
- Wu, W.L.; Hsiao, E.Y.; Yan, Z.; Mazmanian, S.K.; Patterson, P.H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 2017, 62, 11–23. [Google Scholar] [CrossRef]
- Osman, H.C.; Moreno, R.; Rose, D.; Rowland, M.E.; Ciernia, A.V.; Ashwood, P. Impact of maternal immune activation and sex on placental and fetal brain cytokine and gene expression profiles in a preclinical model of neurodevelopmental disorders. J. Neuroinflamm. 2024, 21, 118. [Google Scholar] [CrossRef]
- Tartaglione, A.M.; Villani, A.; Ajmone-Cat, M.A.; Minghetti, L.; Ricceri, L.; Pazienza, V.; De Simone, R.; Calamandrei, G. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl. Psychiatry 2022, 12, 384. [Google Scholar] [CrossRef]
- Al-Haddad, B.J.S.; Oler, E.; Armistead, B.; Elsayed, N.A.; Weinberger, D.R.; Bernier, R.; Burd, I.; Kapur, R.; Jacobsson, B.; Wang, C.; et al. The fetal origins of mental illness. Am. J. Obstet. Gynecol. 2019, 221, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.; Sébire, H.; Brochu, M.E.; Briota, S.; Sarret, P.; Sébire, G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav. Immun. 2012, 26, 1331–1339. [Google Scholar] [CrossRef]
- Girard, S.; Tremblay, L.; Lepage, M.; Sébire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef]
- Girard, S.; Tremblay, L.; Lepage, M.; Sebire, G. Early detection of placental inflammation by MRI enabling protection by clinically relevant IL-1Ra administration. Am. J. Obstet. Gynecol. 2012, 206, 358.e351–358.e359. [Google Scholar] [CrossRef] [PubMed]
- Habelrih, T.; Tremblay, D.; Di Battista, E.; Hou, X.; Reuben, A.; Ferri, B.; Loiselle, S.E.; Côté, F.; Abram, P.; Lubell, W.D.; et al. Pharmacodynamic characterization of rytvela, a novel allosteric anti-inflammatory therapeutic, to prevent preterm birth and improve fetal and neonatal outcomes. Am. J. Obstet. Gynecol. 2023, 228, 467.e1–467.e16. [Google Scholar] [CrossRef]
- Nadeau-Vallée, M.; Chin, P.Y.; Belarbi, L.; Brien, M.; Pundir, S.; Berryer, M.H.; Beaudry-Richard, A.; Madaan, A.; Sharkey, D.J.; Lupien-Meilleur, A.; et al. Antenatal Suppression of IL-1 Protects against Inflammation-Induced Fetal Injury and Improves Neonatal and Developmental Outcomes in Mice. J. Immunol. 2017, 198, 2047–2062. [Google Scholar] [CrossRef]
- Raia-Barjat, T.; Digonnet, M.; Giraud, A.; Ayash, T.; Vancolen, S.; Benharouga, M.; Chauleur, C.; Alfaidy, N.; Sébire, G. Animal Models of Chorioamnionitis: Considerations for Translational Medicine. Biomedicines 2022, 10, 811. [Google Scholar] [CrossRef]
- Andrade, E.B.; Magalhães, A.; Puga, A.; Costa, M.; Bravo, J.; Portugal, C.C.; Ribeiro, A.; Correia-Neves, M.; Faustino, A.; Firon, A.; et al. A mouse model reproducing the pathophysiology of neonatal group B streptococcal infection. Nat. Commun. 2018, 9, 3138. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 2003, 23, 297–302. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Zhang, L.; Dai, X.; Chen, H.; Xie, Q. Anti-tumor necrosis factor-α therapy may not be safe during pregnancy in women with inflammatory bowel disease: An updated meta-analysis and systematic review. BMC Pregnancy Childbirth 2024, 24, 251. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, E.K.; Lee, K.Y.; Kim, H.S. TNF-α antagonist attenuates systemic lipopolysaccharide-induced brain white matter injury in neonatal rats. BMC Neurosci. 2019, 20, 45. [Google Scholar] [CrossRef]
- Dai, F.F.; Hu, M.; Zhang, Y.W.; Zhu, R.H.; Chen, L.P.; Li, Z.D.; Huang, Y.J.; Hu, W.; Cheng, Y.X. TNF-α/anti-TNF-α drugs and its effect on pregnancy outcomes. Expert. Rev. Mol. Med. 2022, 24, e26. [Google Scholar] [CrossRef] [PubMed]
- Farias-Jofre, M.; Romero, R.; Galaz, J.; Xu, Y.; Miller, D.; Garcia-Flores, V.; Arenas-Hernandez, M.; Winters, A.D.; Berkowitz, B.A.; Podolsky, R.H.; et al. Blockade of IL-6R prevents preterm birth and adverse neonatal outcomes. EBioMedicine 2023, 98, 104865. [Google Scholar] [CrossRef]
- Baines, K.J.; West, R.C. Sex differences in innate and adaptive immunity impact fetal, placental, and maternal health†. Biol. Reprod. 2023, 109, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Shook, L.L.; Batorsky, R.E.; De Guzman, R.M.; McCrea, L.T.; Brigida, S.M.; Horng, J.E.; Sheridan, S.D.; Kholod, O.; Cook, A.M.; Li, J.Z.; et al. Maternal SARS-CoV-2 impacts fetal placental macrophage programs and placenta-derived microglial models of neurodevelopment. J. Neuroinflamm. 2024, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, D.; Murgas, P.; Riquelme, E.; Yang, G.; Cancino, G.I. Consequences of Viral Infection and Cytokine Production During Pregnancy on Brain Development in Offspring. Front. Immunol. 2022, 13, 816619. [Google Scholar] [CrossRef]
- Ba, F.; Li, H.; Ding, S.; Guo, T.; Zhang, J.; Sun, Y. Zika Virus-Related Birth Defects and Neurological Complications: A Systematic Review and Meta-Analysis. Rev. Med. Virol. 2025, 35, e70019. [Google Scholar] [CrossRef]
- de Oliveira Campos Amaral, A.; de Oliveira Pache de Faria, A.; Carvalho, F.R.; Dalcastel, L.A.B.; Almeida, S.S.A.; Fernandes, A.R.; Velarde, L.G.C.; Oliveira, S.A.; Cardoso, C.A.A.; Miterhof, M.; et al. Association between microcephaly and hearing disorders in children exposed or suspected of exposure to the Zika virus during the intrauterine period. Eur. J. Pediatr. 2024, 184, 78. [Google Scholar] [CrossRef]
- Aagaard, K.M.; Lahon, A.; Suter, M.A.; Arya, R.P.; Seferovic, M.D.; Vogt, M.B.; Hu, M.; Stossi, F.; Mancini, M.A.; Harris, R.A.; et al. Primary Human Placental Trophoblasts are Permissive for Zika Virus (ZIKV) Replication. Sci. Rep. 2017, 7, 41389. [Google Scholar] [CrossRef]
- Arruda, L.V.; Salomão, N.G.; Alves, F.A.V.; Rabelo, K. The Innate Defense in the Zika-Infected Placenta. Pathogens 2022, 11, 1410. [Google Scholar] [CrossRef]
- Oseghale, O.; Vlahos, R.; O’Leary, J.J.; Brooks, R.D.; Brooks, D.A.; Liong, S.; Selemidis, S. Influenza Virus Infection during Pregnancy as a Trigger of Acute and Chronic Complications. Viruses 2022, 14, 2729. [Google Scholar] [CrossRef] [PubMed]
- Nijman, M.; van der Meeren, L.E.; Nikkels, P.G.J.; Stegeman, R.; Breur, J.; Jansen, N.J.G.; Ter Heide, H.; Steenhuis, T.J.; de Heus, R.; Bekker, M.N.; et al. Placental Pathology Contributes to Impaired Volumetric Brain Development in Neonates with Congenital Heart Disease. J. Am. Heart Assoc. 2024, 13, e033189. [Google Scholar] [CrossRef] [PubMed]
- Segar, D.E.; Zhang, J.; Yan, K.; Reid, A.; Frommelt, M.; Cohen, S. The Relationship Between Placental Pathology and Neurodevelopmental Outcomes in Complex Congenital Heart Disease. Pediatr. Cardiol. 2023, 44, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Tsuchiya, K.J.; Yaguchi, C.; Furuta-Isomura, N.; Horikoshi, Y.; Matsumoto, M.; Suzuki, M.; Oda, T.; Kawai, K.; Itoh, T.; et al. Placental pathology predicts infantile neurodevelopment. Sci. Rep. 2022, 12, 2578. [Google Scholar] [CrossRef]
- Yaguchi, C.; Ueda, M.; Mizuno, Y.; Fukuchi, C.; Matsumoto, M.; Furuta-Isomura, N.; Itoh, H. Association of Placental Pathology with Physical and Neuronal Development of Infants: A Narrative Review and Reclassification of the Literature by the Consensus Statement of the Amsterdam Placental Workshop Group. Nutrients 2024, 16, 1786. [Google Scholar] [CrossRef]
- Chen, S.; Shenoy, A. Placental Pathology and the Developing Brain. Semin. Pediatr. Neurol. 2022, 42, 100975. [Google Scholar] [CrossRef]
- Spinillo, A.; Dominoni, M.; Caporali, C.; Olivieri, I.; La Piana, R.; Longo, S.; Cesari, S.; Fiandrino, G.; Orcesi, S.; Gardella, B. Placental Histological Features and Neurodevelopmental Outcomes at Two Years in Very-Low-Birth-Weight Infants. Pediatr. Neurol. 2021, 120, 63–70. [Google Scholar] [CrossRef]
- Couture, C.; Caron, M.; St-Onge, P.; Brien, M.E.; Sinnett, D.; Dal Soglio, D.; Girard, S. Identification of divergent placental profiles in clinically distinct pregnancy complications revealed by the transcriptome. Placenta 2024, 154, 184–192. [Google Scholar] [CrossRef]
- Orton, J.; Doyle, L.W.; Tripathi, T.; Boyd, R.; Anderson, P.J.; Spittle, A. Early developmental intervention programmes provided post hospital discharge to prevent motor and cognitive impairment in preterm infants. Cochrane Database Syst. Rev. 2024, 2, CD005495. [Google Scholar] [CrossRef]
- Brien, M.E.; Gaudreault, V.; Hughes, K.; Hayes, D.J.L.; Heazell, A.E.P.; Girard, S. A Systematic Review of the Safety of Blocking the IL-1 System in Human Pregnancy. J. Clin. Med. 2021, 11, 225. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervantes, E.M.; Girard, S. Placental Inflammation in Preterm Premature Rupture of Membranes and Risk of Neurodevelopmental Disorders. Cells 2025, 14, 965. https://doi.org/10.3390/cells14130965
Cervantes EM, Girard S. Placental Inflammation in Preterm Premature Rupture of Membranes and Risk of Neurodevelopmental Disorders. Cells. 2025; 14(13):965. https://doi.org/10.3390/cells14130965
Chicago/Turabian StyleCervantes, Elizabeth Marie, and Sylvie Girard. 2025. "Placental Inflammation in Preterm Premature Rupture of Membranes and Risk of Neurodevelopmental Disorders" Cells 14, no. 13: 965. https://doi.org/10.3390/cells14130965
APA StyleCervantes, E. M., & Girard, S. (2025). Placental Inflammation in Preterm Premature Rupture of Membranes and Risk of Neurodevelopmental Disorders. Cells, 14(13), 965. https://doi.org/10.3390/cells14130965