
Duchenne Muscular Dystrophy Patient iPSCs—Derived Skeletal Muscle Organoids Exhibit a Developmental Delay in Myogenic Progenitor Maturation

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Human-Induced Pluripotent Stem Cell (iPSC) Lines
2.2. hiPSC Culture
2.3. Skeletal Muscle Organoid Differentiation
2.4. scRNA-seq Sample Preparation and Sequencing
2.5. Data Analysis Pipeline
2.5.1. Raw Data Processing with Cell Ranger
2.5.2. Seurat Data Analysis
2.5.3. Quality Control for Organoid-Derived Datasets
2.5.4. Integrative Seurat Analysis
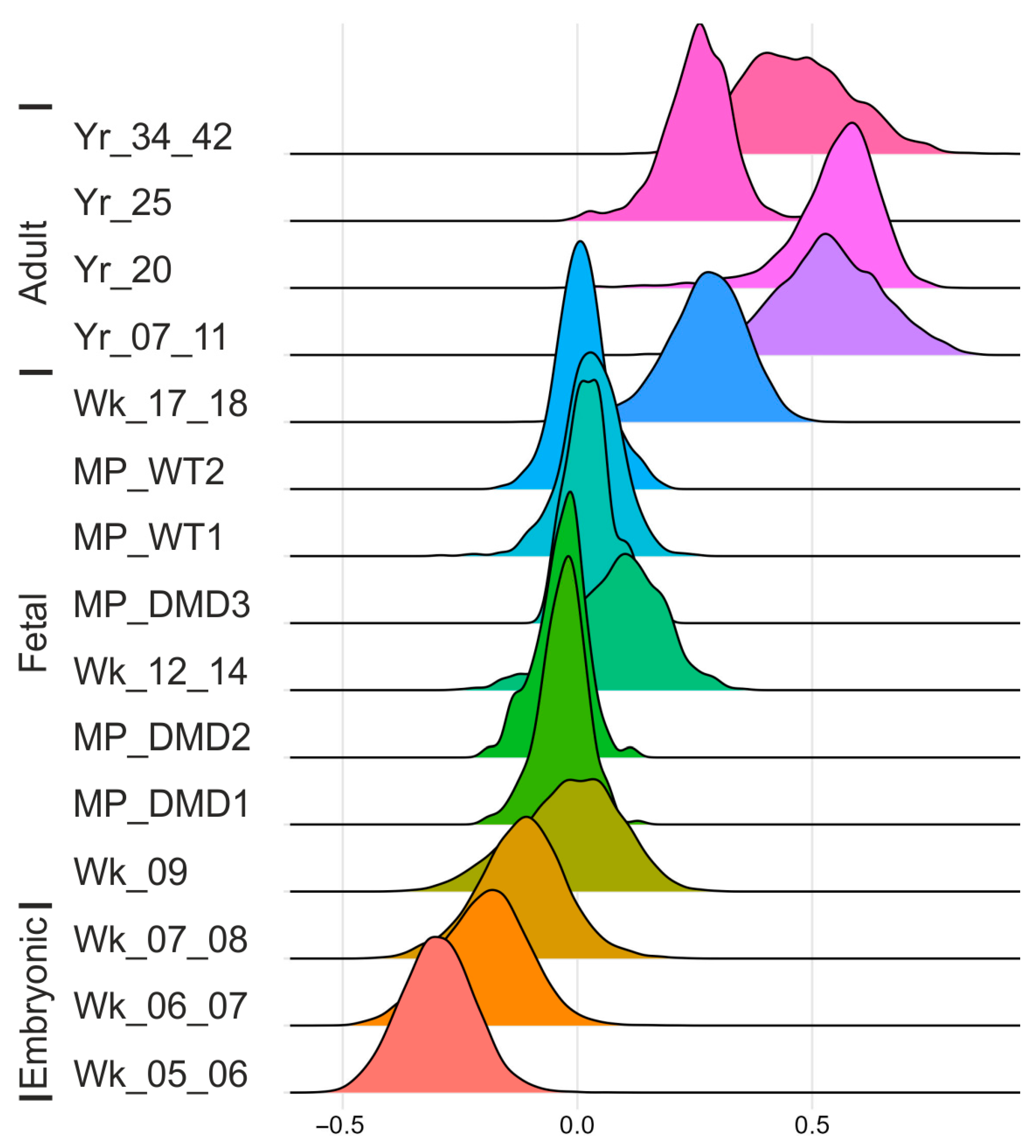
2.5.5. Developmental Score Analysis
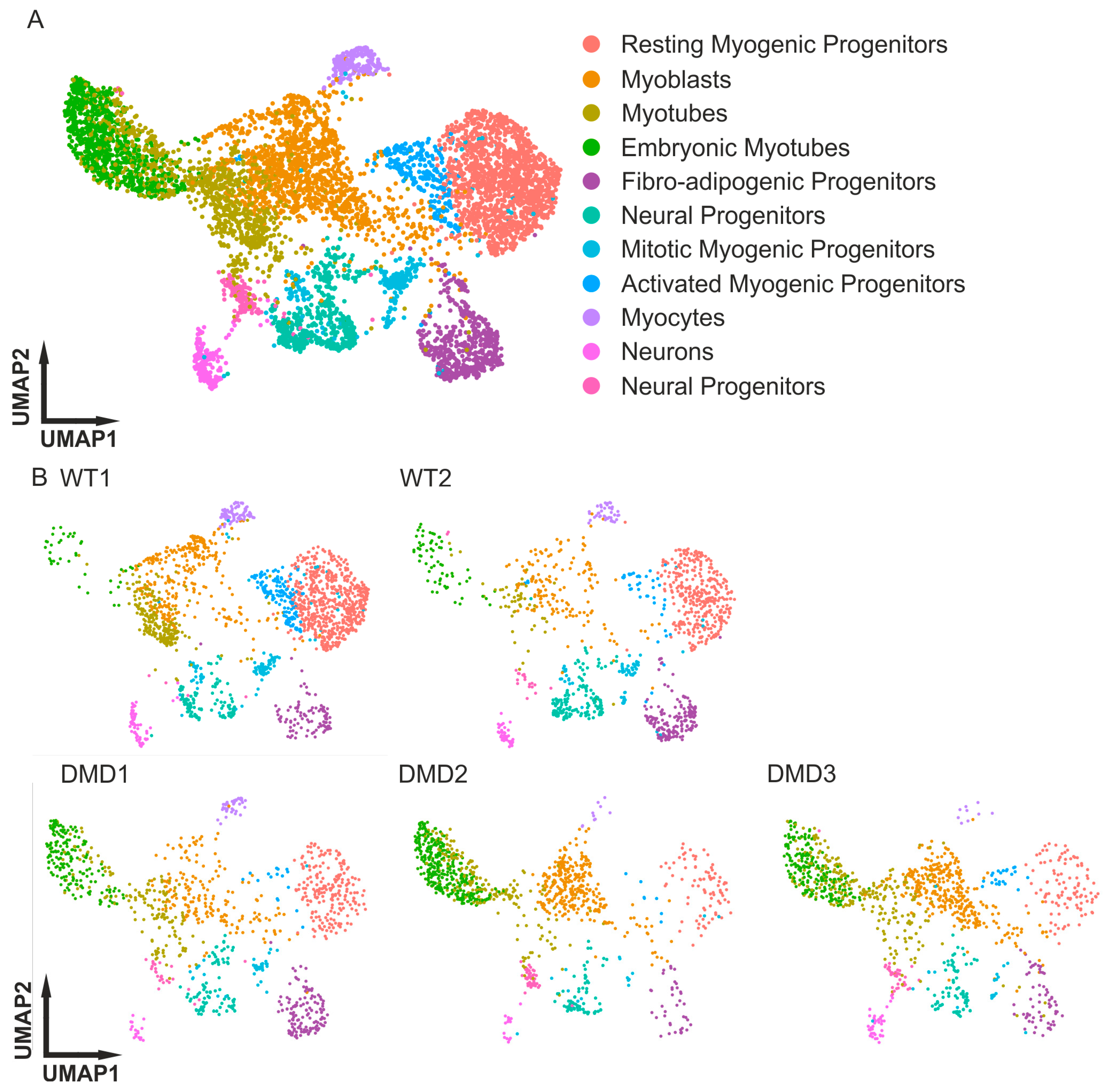
3. Results
3.1. Quality Control for Single-Cell Data
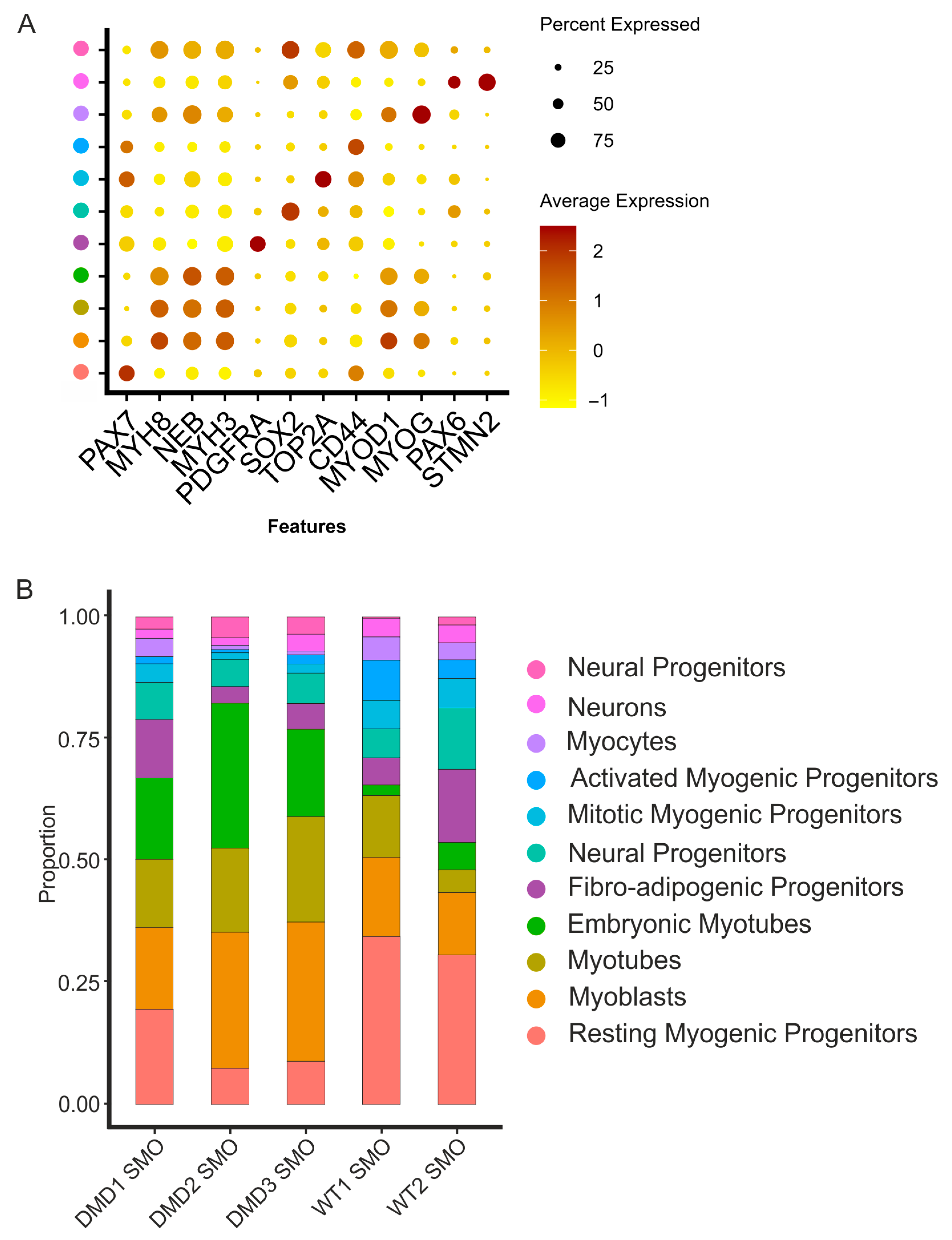
3.2. Integrative Comparison of the 3D Skeletal Muscle Organoid Systems
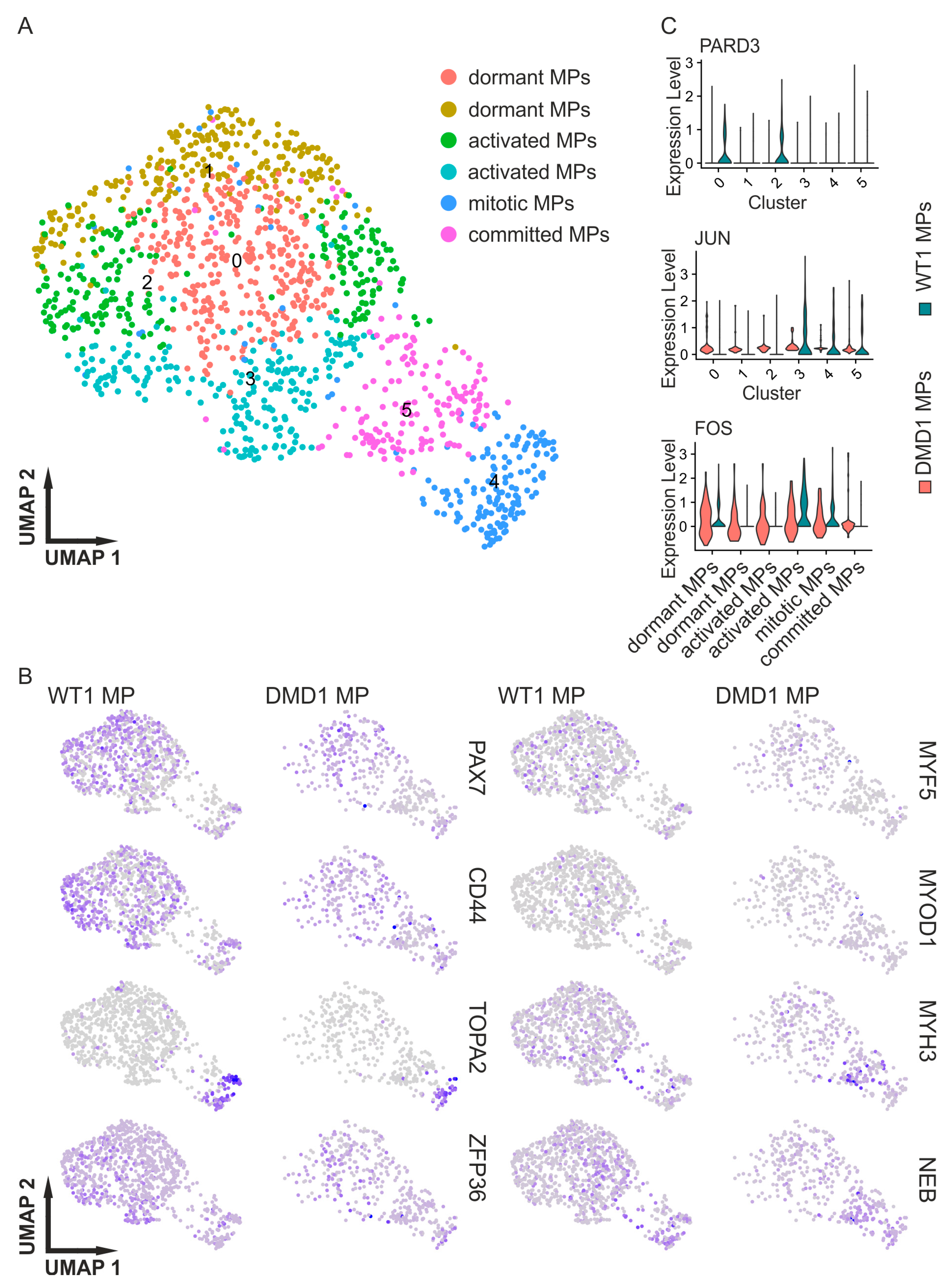
3.3. DMD Myogenic Progenitors Exhibit an Altered Transcriptional Profile
4. Discussion
4.1. Integrative Analysis Implementing the Robust Reproducibility of the SMO Model in Healthy and Diseased Cell Lines
4.2. Fibro-Adipogenic Progenitors
4.3. Myogenic Progenitors
4.4. Myogenic Maturation Score
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 Version of the Gene Table of Monogenic Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2017, 27, 1152–1183. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Bonne, G.; Rivier, F.; Hamroun, D. The 2022 Version of the Gene Table of Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2021, 31, 1313–1357. [Google Scholar] [CrossRef] [PubMed]
- Huml, R.A. Muscular Dystrophy: Historical Background and Types. In Muscular Dystrophy; Springer: Cham, Switzerland, 2015; Volume 31, pp. 5–7. [Google Scholar] [CrossRef]
- Med, D.D.-A.G. Undefined Recherches Sur La Paralysie Musculaire Pseudo Hypertrophique Ou Paralysie Myo-Sclerosique; P. Asselin: Paris, Frence, 1868. [Google Scholar]
- Yiu, E.; Kornberg, A. Duchenne Muscular Dystrophy. Neurol. India 2008, 56, 236. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Swandulla, D. Complexity of Skeletal Muscle Degeneration: Multi-Systems Pathophysiology and Organ Crosstalk in Dystrophinopathy. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 1813–1839. [Google Scholar] [CrossRef]
- Serrano, A.L.; Mann, C.J.; Vidal, B.; Ardite, E.; Perdiguero, E.; Muñoz-Cánoves, P. Cellular and Molecular Mechanisms Regulating Fibrosis in Skeletal Muscle Repair and Disease. Curr. Top. Dev. Biol. 2011, 96, 167–201. [Google Scholar]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing Skeletal Muscle: Regenerative Potential of Skeletal Muscle Stem Cells. J Clin. Investig. 2010, 120, 11–19. [Google Scholar] [CrossRef]
- Schmidt, M.; Schüler, S.C.; Hüttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult Stem Cells at Work: Regenerating Skeletal Muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and Extrinsic Mechanisms Regulating Satellite Cell Function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Günther, S.; Kim, J.; Kostin, S.; Lepper, C.; Fan, C.-M.; Braun, T. Myf5-Positive Satellite Cells Contribute to Pax7-Dependent Long-Term Maintenance of Adult Muscle Stem Cells. Cell Stem Cell 2013, 13, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Geles, K.G.; Paik, J.-H.; DePinho, R.A.; Tjian, R. Codependent Activators Direct Myoblast-Specific MyoD Transcription. Dev. Cell 2008, 15, 534–546. [Google Scholar] [CrossRef]
- Judson, R.N.; Quarta, M.; Oudhoff, M.J.; Soliman, H.; Yi, L.; Chang, C.K.; Loi, G.; Vander Werff, R.; Cait, A.; Hamer, M.; et al. Inhibition of Methyltransferase Setd7 Allows the In Vitro Expansion of Myogenic Stem Cells with Improved Therapeutic Potential. Cell Stem Cell 2018, 22, 177–190.e7. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.G. Does the Pathogenic Sequence of Skeletal Muscle Degeneration in Duchenne Muscular Dystrophy Begin and End with Unrestrained Satellite Cell Activation? Muscles 2022, 1, 75–81. [Google Scholar] [CrossRef]
- Kottlors, M.; Kirschner, J. Elevated Satellite Cell Number in Duchenne Muscular Dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin Expression in Muscle Stem Cells Regulates Their Polarity and Asymmetric Division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Yamashita, K.; Suzuki, A.; Satoh, Y.; Ide, M.; Amano, Y.; Masuda-Hirata, M.; Hayashi, Y.K.; Hamada, K.; Ogata, K.; Ohno, S. The 8th and 9th Tandem Spectrin-like Repeats of Utrophin Cooperatively Form a Functional Unit to Interact with Polarity-Regulating Kinase PAR-1b. Biochem. Biophys. Res. Commun. 2010, 391, 812–817. [Google Scholar] [CrossRef]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT Signaling Stimulates Adult Satellite Cell Function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy-Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and FACS-Mediated Purification of PAX3+/PAX7+ Skeletal Muscle Precursors from Human Pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of Pluripotent Stem Cells to Muscle Fiber to Model Duchenne Muscular Dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of Human Muscle Fibers and Satellite-like Cells from Human Pluripotent Stem Cells in Vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.R.; Hiserodt, J.; Paras, K.; Fujiwara, W.; Eskin, A.; Jan, M.; Xi, H.; Young, C.S.; Evseenko, D.; Nelson, S.F.; et al. ERBB3 and NGFR Mark a Distinct Skeletal Muscle Progenitor Cell in Human Development and HPSCs. Nat. Cell Biol. 2018, 20, 46–57. [Google Scholar] [CrossRef]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.P.; Stanford, W.L.; Skerjanc, I.S. Derivation and Expansion of PAX7-Positive Muscle Progenitors from Human and Mouse Embryonic Stem Cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef]
- Xi, H.; Young, C.S.; Pyle, A.D. Generation of PAX7 Reporter Cells to Investigate Skeletal Myogenesis from Human Pluripotent Stem Cells. STAR Protoc. 2020, 1, 100158. [Google Scholar] [CrossRef]
- Cuny, G.D.; Yu, P.B.; Laha, J.K.; Xing, X.; Liu, J.F.; Lai, C.S.; Deng, D.Y.; Sachidanandan, C.; Bloch, K.D.; Peterson, R.T. Structure–Activity Relationship Study of Bone Morphogenetic Protein (BMP) Signaling Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. [Google Scholar] [CrossRef]
- Gouti, M.; Tsakiridis, A.; Wymeersch, F.J.; Huang, Y.; Kleinjung, J.; Wilson, V.; Briscoe, J. In Vitro Generation of Neuromesodermal Progenitors Reveals Distinct Roles for Wnt Signalling in the Specification of Spinal Cord and Paraxial Mesoderm Identity. PLoS Biol. 2014, 12, e1001937. [Google Scholar] [CrossRef]
- Tanoury, Z.A.; Rao, J.; Tassy, O.; Gobert, B.; Gapon, S.; Garnier, J.M.; Wagner, E.; Hick, A.; Hall, A.; Gussoni, E.; et al. Differentiation of the Human PAX7-Positive Myogenic Precursors/Satellite Cell Lineage in Vitro. Development 2020, 147, dev187344. [Google Scholar] [CrossRef]
- Mashinchian, O.; Pisconti, A.; Le Moal, E.; Bentzinger, C.F. The Muscle Stem Cell Niche in Health and Disease. Curr. Top Dev. Biol. 2018, 126, 23–65. [Google Scholar] [CrossRef]
- Relaix, F.; Bencze, M.; Borok, M.J.; Der Vartanian, A.; Gattazzo, F.; Mademtzoglou, D.; Perez-Diaz, S.; Prola, A.; Reyes-Fernandez, P.C.; Rotini, A.; et al. Perspectives on Skeletal Muscle Stem Cells. Nat. Commun. 2021, 12, 692. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in Vitro Model of Human Development and Disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 6194. [Google Scholar] [CrossRef] [PubMed]
- Faustino Martins, J.M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 26, 172–186.e6. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human IPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef]
- Mavrommatis, L.; Jeong, H.-W.; Gomez-Giro, G.; Stehling, M.; Kienitz, M.-C.; Psathaki, O.E.; Bixel, M.G.; Morosan-Puopolo, G.; Gerovska, D.; Araúzo-Bravo, M.J.; et al. Human Skeletal Muscle Organoids Model Fetal Myogenesis and Sustain Uncommitted PAX7 Myogenic Progenitors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mavrommatis, L.; Jeong, H.-W.; Kindler, U.; Gomez-Giro, G.; Kienitz, M.-C.; Stehling, M.; Psathaki, O.E.; Zeuschner, D.; Bixel, M.G.; Han, D.; et al. Human Skeletal Muscle Organoids Model Fetal Myogenesis and Sustain Uncommitted PAX7 Myogenic Progenitors. eLife 2023, 12, RP87081. [Google Scholar] [CrossRef]
- Pinton, L.; Khedr, M.; Lionello, V.M.; Sarcar, S.; Maffioletti, S.M.; Dastidar, S.; Negroni, E.; Choi, S.W.; Khokhar, N.; Bigot, A.; et al. 3D Human Induced Pluripotent Stem Cell–Derived Bioengineered Skeletal Muscles for Tissue, Disease and Therapy Modeling. Nat. Protoc. 2023, 18, 1337–1376. [Google Scholar] [CrossRef]
- Shin, M.K.; Bang, J.S.; Lee, J.E.; Tran, H.D.; Park, G.; Lee, D.R.; Jo, J. Generation of Skeletal Muscle Organoids from Human Pluripotent Stem Cells to Model Myogenesis and Muscle Regeneration. Int. J. Mol. Sci. 2022, 23, 5108. [Google Scholar] [CrossRef]
- Shahriyari, M.; Islam, M.R.; Sakib, S.M.; Rinn, M.; Rika, A.; Krüger, D.; Kaurani, L.; Gisa, V.; Winterhoff, M.; Anandakumar, H.; et al. Engineered Skeletal Muscle Recapitulates Human Muscle Development, Regeneration and Dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 3106–3121. [Google Scholar] [CrossRef]
- Cairns, D.M.; Sato, M.E.; Lee, P.G.; Lassar, A.B.; Zeng, L. A Gradient of Shh Establishes Mutually Repressing Somitic Cell Fates Induced by Nkx3.2 and Pax3. Dev. Biol. 2008, 323, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.D.; D’Antonio, M.; Benaglio, P.; Williams, R.; Hashem, S.I.; Schuldt, B.M.; DeBoever, C.; Arias, A.D.; Garcia, M.; Nelson, B.C.; et al. IPSCORE: A Resource of 222 IPSC Lines Enabling Functional Characterization of Genetic Variation across a Variety of Cell Types. Stem Cell Rep. 2017, 8, 1086–1100. [Google Scholar] [CrossRef] [PubMed]
- Dorn, I.; Klich, K.; Arauzo-Bravo, M.J.; Radstaak, M.; Santourlidis, S.; Ghanjati, F.; Radke, T.F.; Psathaki, O.E.; Hargus, G.; Kramer, J.; et al. Erythroid Differentiation of Human Induced Pluripotent Stem Cells Is Independent of Donor Cell Type of Origin. Haematologica 2015, 100, 32–41. [Google Scholar] [CrossRef]
- Kindler, U.; Zaehres, H.; Mavrommatis, L. Generation of Skeletal Muscle Organoids from Human Pluripotent Stem Cells. Bio Protoc. 2024, 14, e4984. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Van Den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; Van Oudenaarden, A. Single-Cell Sequencing Reveals Dissociation-Induced Gene Expression in Tissue Subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef]
- Rubenstein, A.B.; Smith, G.R.; Raue, U.; Begue, G.; Minchev, K.; Ruf-Zamojski, F.; Nair, V.D.; Wang, X.; Zhou, L.; Zaslavsky, E.; et al. Single-Cell Transcriptional Profiles in Human Skeletal Muscle. Sci. Rep. 2020, 10, 229. [Google Scholar] [CrossRef]
- Xi, H.; Langerman, J.; Sabri, S.; Chien, P.; Young, C.S.; Younesi, S.; Hicks, M.; Gonzalez, K.; Fujiwara, W.; Marzi, J.; et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell 2020, 27, 158–176.e10. [Google Scholar] [CrossRef] [PubMed]
- Barruet, E.; Garcia, S.M.; Striedinger, K.; Wu, J.; Lee, S.; Byrnes, L.; Wong, A.; Xuefeng, S.; Tamaki, S.; Brack, A.S.; et al. Functionally Heterogeneous Human Satellite Cells Identified by Single Cell RNA Sequencing. eLife 2020, 9, e51576. [Google Scholar] [CrossRef] [PubMed]
- Germain, P.L.; Sonrel, A.; Robinson, M.D. PipeComp, a General Framework for the Evaluation of Computational Pipelines, Reveals Performant Single Cell RNA-Seq Preprocessing Tools. Genome Biol. 2020, 21, 227. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A Practical Guide to Single-Cell RNA-Sequencing for Biomedical Research and Clinical Applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating Single-Cell Analysis with Bioconductor. Nat. Methods 2019, 17, 137–145. [Google Scholar] [CrossRef]
- Lun, A.T.; McCarthy, D.J.; Marioni, J.C.; Ji, H.; du Verle, D.; Rausell, A.; Descartes, P. A Step-by-Step Workflow for Low-Level Analysis of Single-Cell RNA-Seq Data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Mournetas, V.; Massouridès, E.; Dupont, J.B.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis Modelled by Human Pluripotent Stem Cells: A Multi-Omic Study of Duchenne Myopathy Early Onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef]
- Saleh, K.K.; Xi, H.; Switzler, C.; Skuratovsky, E.; Romero, M.A.; Chien, P.; Gibbs, D.; Gane, L.; Hicks, M.R.; Spencer, M.J.; et al. Single Cell Sequencing Maps Skeletal Muscle Cellular Diversity as Disease Severity Increases in Dystrophic Mouse Models. iScience 2022, 25, 105415. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Homma, S.T.; Wang, Y.; Smith, G.R.; Ruf-Zamojski, F.; Sealfon, S.C.; Zhou, L. Diverse Effector and Regulatory Functions of Fibro/Adipogenic Progenitors during Skeletal Muscle Fibrosis in Muscular Dystrophy. iScience 2023, 26, 105775. [Google Scholar] [CrossRef]
- Prüller, J.; Mannhardt, I.; Eschenhagen, T.; Zammit, P.S.; Figeac, N. Satellite Cells Delivered in Their Niche Efficiently Generate Functional Myotubes in Three-Dimensional Cell Culture. PLoS ONE 2018, 13, e0202574. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. A Pax3/Pax7-Dependent Population of Skeletal Muscle Progenitor Cells. Nature 2005, 435, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 Is Required for the Specification of Myogenic Satellite Cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Nesmith, A.P.; Wagner, M.A.; Pasqualini, F.S.; O’Connor, B.B.; Pincus, M.J.; August, P.R.; Parker, K.K. A Human in Vitro Model of Duchenne Muscular Dystrophy Muscle Formation and Contractility. J. Cell Biol. 2016, 215, 47–56. [Google Scholar] [CrossRef]
- Feige, P.; Brun, C.E.; Ritso, M.; Rudnicki, M.A. Orienting Muscle Stem Cells for Regeneration in Homeostasis, Aging, and Disease. Cell Stem Cell 2018, 23, 653–664. [Google Scholar] [CrossRef]
- Heslop, L.; Morgan, J.E.; Partridge, T.A. Evidence for a Myogenic Stem Cell That Is Exhausted in Dystrophic Muscle. J. Cell Sci. 2000, 113, 2299–2308. [Google Scholar] [CrossRef]
- Zammit, P.S.; Golding, J.P.; Nagata, Y.; Hudon, V.; Partridge, T.A.; Beauchamp, J.R. Muscle Satellite Cells Adopt Divergent Fates a Mechanism for Self-Renewal? J. Cell Biol. 2004, 166, 347–357. [Google Scholar] [CrossRef]
- Theret, M.; Rossi, F.M.V.; Contreras, O. Evolving Roles of Muscle-Resident Fibro-Adipogenic Progenitors in Health, Regeneration, Neuromuscular Disorders, and Aging. Front. Physiol. 2021, 12, 673404. [Google Scholar] [CrossRef]
- Mázala, D.A.G.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-β–Driven Muscle Degeneration and Failed Regeneration Underlie Disease Onset in a DMD Mouse Model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef]
- Moratal, C.; Arrighi, N.; Dechesne, C.A.; Dani, C. Control of Muscle Fibro-Adipogenic Progenitors by Myogenic Lineage Is Altered in Aging and Duchenne Muscular Dystrophy. Cell. Physiol. Biochem. 2019, 53, 1029–1045. [Google Scholar] [CrossRef]
- Rayagiri, S.S.; Ranaldi, D.; Raven, A.; Mohamad Azhar, N.I.F.; Lefebvre, O.; Zammit, P.S.; Borycki, A.G. Basal Lamina Remodeling at the Skeletal Muscle Stem Cell Niche Mediates Stem Cell Self-Renewal. Nat. Commun. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Marg, A.; Escobar, H.; Karaiskos, N.; Grunwald, S.A.; Metzler, E.; Kieshauer, J.; Sauer, S.; Pasemann, D.; Malfatti, E.; Mompoint, D.; et al. Human Muscle-Derived CLEC14A-Positive Cells Regenerate Muscle Independent of PAX7. Nat. Commun. 2019, 10, 5776. [Google Scholar] [CrossRef] [PubMed]
- Mavrommatis, L.; Zaben, A.; Kindler, U.; Kienitz, M.-C.; Dietz, J.; Jeong, H.-W.; Böhme, P.; Brand-Saberi, B.; Vorgerd, M.; Zaehres, H. CRISPR/Cas9 Genome Editing in LGMD2A/R1 Patient-Derived Induced Pluripotent Stem and Skeletal Muscle Progenitor Cells. Stem Cells Int. 2023, 2023, 9246825. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The Protein Product of the Duchenne Muscular Dystrophy Locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Johnson, E.K.; Li, B.; Yoon, J.H.; Flanigan, K.M.; Martin, P.T.; Ervasti, J.; Montanaro, F. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE. 2013, 8, e73224. [Google Scholar] [CrossRef]
- Silver, J.S.; Günay, K.A.; Cutler, A.A.; Vogler, T.O.; Brown, T.E.; Pawlikowski, B.T.; Bednarski, O.J.; Bannister, K.L.; Rogowski, C.J.; McKay, A.G.; et al. Injury-Mediated Stiffening Persistently Activates Muscle Stem Cells through YAP and TAZ Mechanotransduction. Sci. Adv. 2021, 7, 4501–4513. [Google Scholar] [CrossRef]
- Haroon, M.; Klein-Nulend, J.; Bakker, A.D.; Jin, J.; Seddiqi, H.; Offringa, C.; de Wit, G.M.J.; Le Grand, F.; Giordani, L.; Liu, K.J.; et al. Myofiber Stretch Induces Tensile and Shear Deformation of Muscle Stem Cells in Their Native Niche. Biophys. J. 2021, 120, 2665–2678. [Google Scholar] [CrossRef]
- Tao, J.; Choudhury, M.I.; Maity, D.; Kim, T.; Sun, S.X.; Fan, C.M. Mechanical Compression Creates a Quiescent Muscle Stem Cell Niche. Commun. Biol. 2023, 6, 43. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | WT1 SMO | WT2 SMO | DMD1 SMO | DMD2 SMO | DMD3 SMO |
|---|---|---|---|---|---|
| Cell Type | |||||
| Neural Progenitors | 0.22% | 1.63% | 2.49% | 4.23% | 3.41% |
| Neurons | 3.62% | 3.51% | 1.75% | 1.44% | 3.32% |
| Myocytes | 4.89% | 3.51% | 3.78% | 0.90% | 0.77% |
| Activated Myogenic Progenitors | 8.18% | 3.76% | 1.48% | 0.63% | 1.88% |
| Mitotic Myogenic Progenitors | 5.82% | 6.16% | 3.78% | 1.35% | 1.96% |
| Neural Progenitors | 5.88% | 12.57% | 7.56% | 5.58% | 6.14% |
| Fibro-Adipogenic Progenitors | 5.60% | 14.97% | 11.90% | 3.42% | 5.20% |
| Embryonic Myotubes | 2.20% | 5.65% | 16.70% | 29.79% | 17.99% |
| Myotubes | 12.74% | 4.62% | 14.02% | 17.19% | 21.74% |
| Myoblasts | 16.36% | 12.75% | 16.79% | 27.81% | 28.56% |
| Resting Myogenic Progenitors | 34.49% | 30.88% | 19.65% | 7.65% | 9.04% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kindler, U.; Mavrommatis, L.; Käppler, F.; Hiluf, D.G.; Heilmann-Heimbach, S.; Marcus, K.; Günther Pomorski, T.; Vorgerd, M.; Brand-Saberi, B.; Zaehres, H. Duchenne Muscular Dystrophy Patient iPSCs—Derived Skeletal Muscle Organoids Exhibit a Developmental Delay in Myogenic Progenitor Maturation. Cells 2025, 14, 1033. https://doi.org/10.3390/cells14131033
Kindler U, Mavrommatis L, Käppler F, Hiluf DG, Heilmann-Heimbach S, Marcus K, Günther Pomorski T, Vorgerd M, Brand-Saberi B, Zaehres H. Duchenne Muscular Dystrophy Patient iPSCs—Derived Skeletal Muscle Organoids Exhibit a Developmental Delay in Myogenic Progenitor Maturation. Cells. 2025; 14(13):1033. https://doi.org/10.3390/cells14131033
Chicago/Turabian StyleKindler, Urs, Lampros Mavrommatis, Franziska Käppler, Dalya Gebrehiwet Hiluf, Stefanie Heilmann-Heimbach, Katrin Marcus, Thomas Günther Pomorski, Matthias Vorgerd, Beate Brand-Saberi, and Holm Zaehres. 2025. "Duchenne Muscular Dystrophy Patient iPSCs—Derived Skeletal Muscle Organoids Exhibit a Developmental Delay in Myogenic Progenitor Maturation" Cells 14, no. 13: 1033. https://doi.org/10.3390/cells14131033
APA StyleKindler, U., Mavrommatis, L., Käppler, F., Hiluf, D. G., Heilmann-Heimbach, S., Marcus, K., Günther Pomorski, T., Vorgerd, M., Brand-Saberi, B., & Zaehres, H. (2025). Duchenne Muscular Dystrophy Patient iPSCs—Derived Skeletal Muscle Organoids Exhibit a Developmental Delay in Myogenic Progenitor Maturation. Cells, 14(13), 1033. https://doi.org/10.3390/cells14131033