
Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Mouse Models
2.3. Airway Responsiveness to Methacholine
2.4. Antibodies and Reagents
2.5. Histopathology, Immunohistochemical and Immunofluorescence Analysis
2.6. Cell Culture and Treatment
2.7. RNA Sequencing
2.8. Western Blotting
2.9. Flow Cytometry
2.10. Quantitative RT-PCR Analysis
2.11. ELISA
2.12. Oxygen Consumption and Glycolytic Rates
2.13. Co-Immunoprecipitation (Co-IP) Assay
2.14. Statistical Analysis
3. Results
3.1. Altered Glucose Metabolism in Asthma
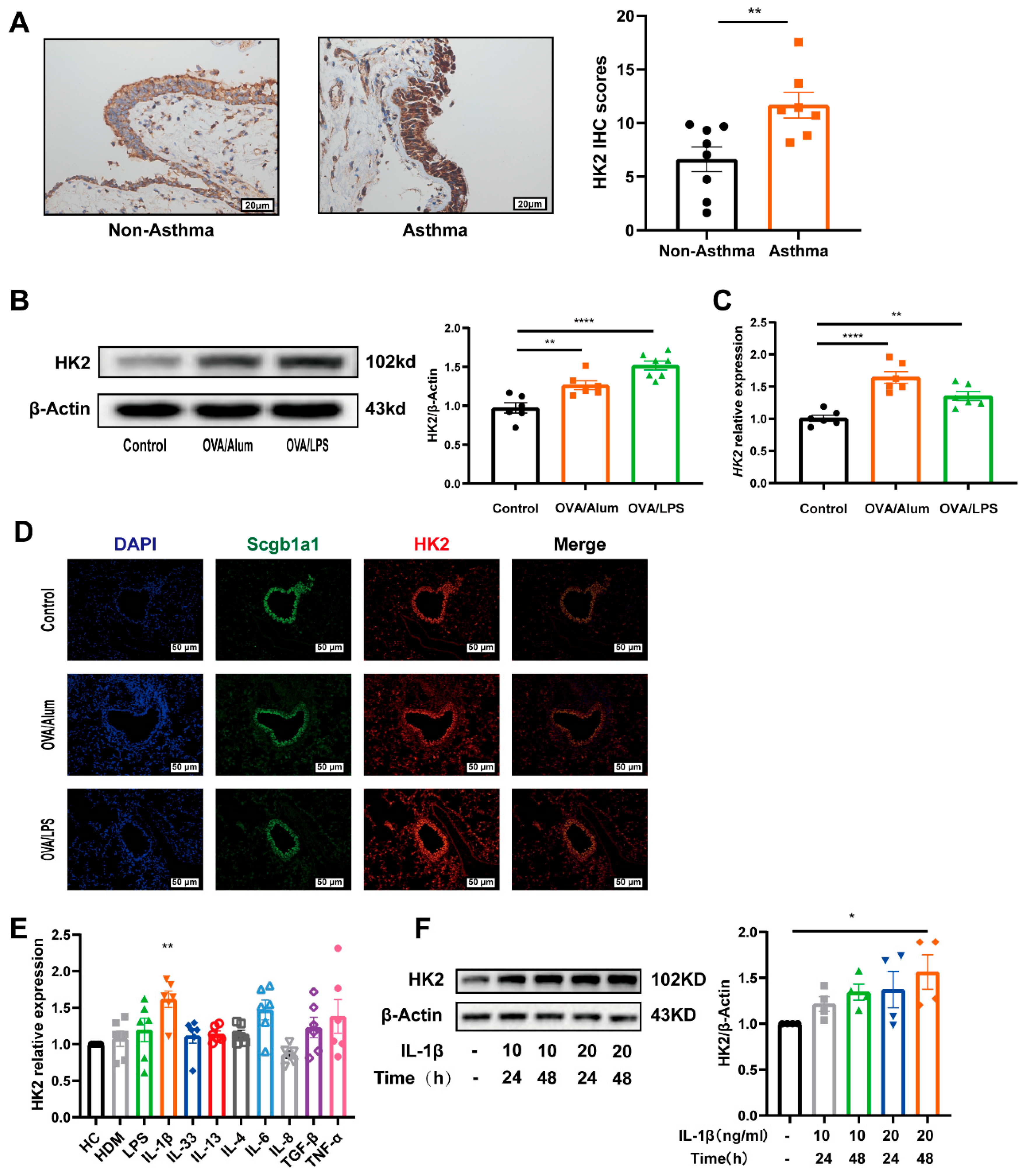
3.2. HK2 Is Upregulated in Asthma
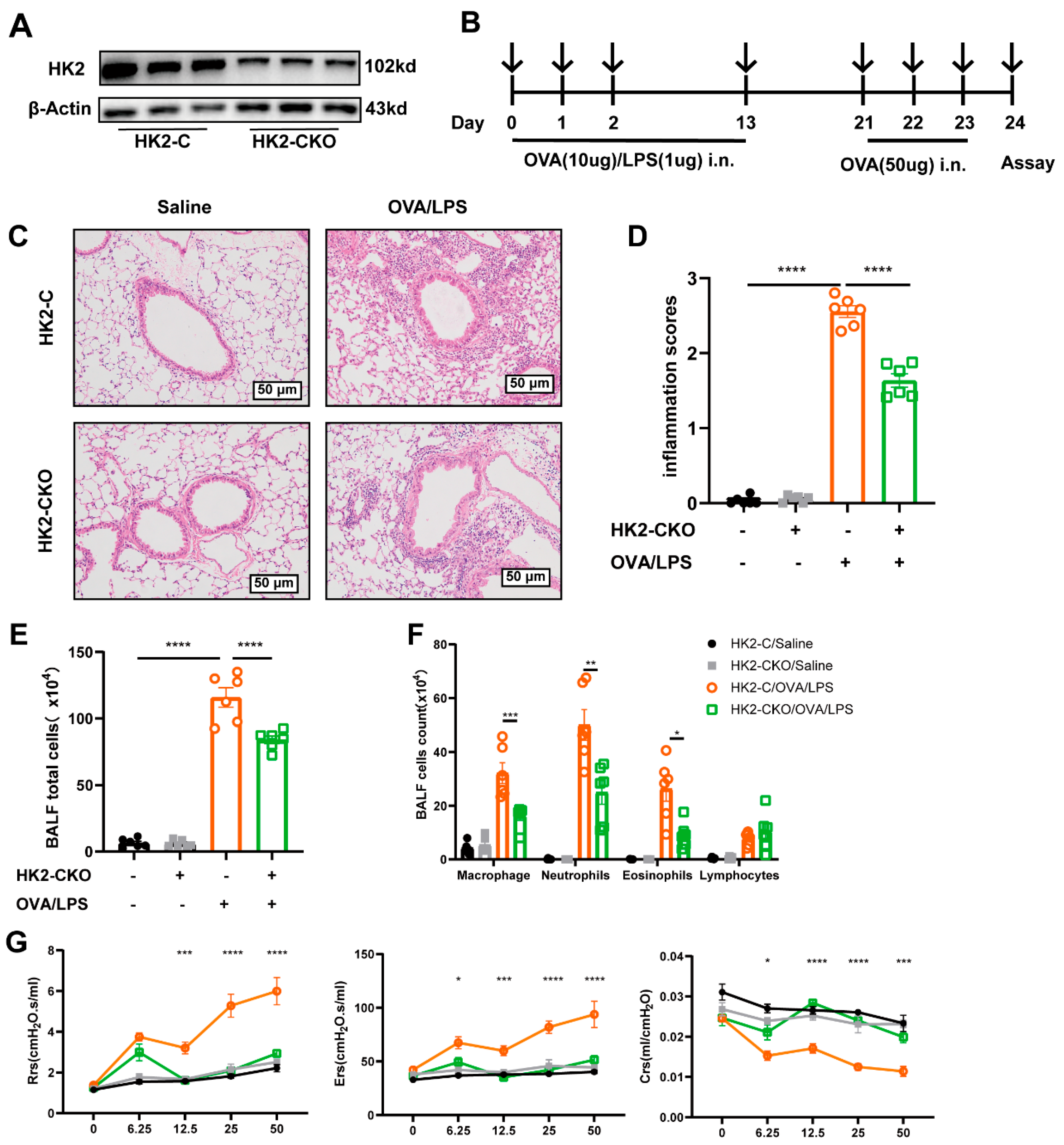
3.3. HK2 Deficiency Ameliorates Inflammation in Asthmatic Mice
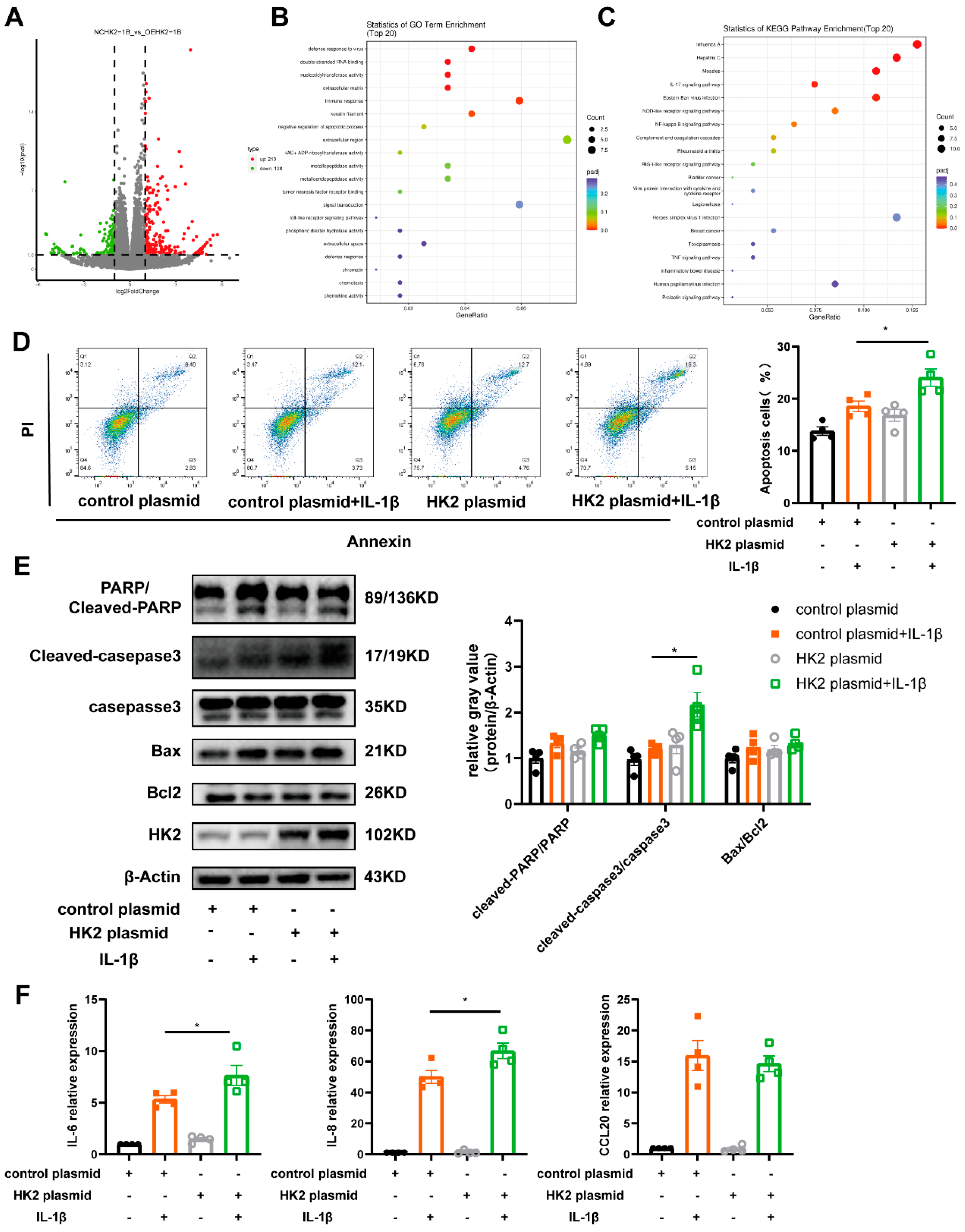
3.4. HK2 Is Involved in Airway Epithelial Cell Apoptosis, Immune Response and Glycolysis
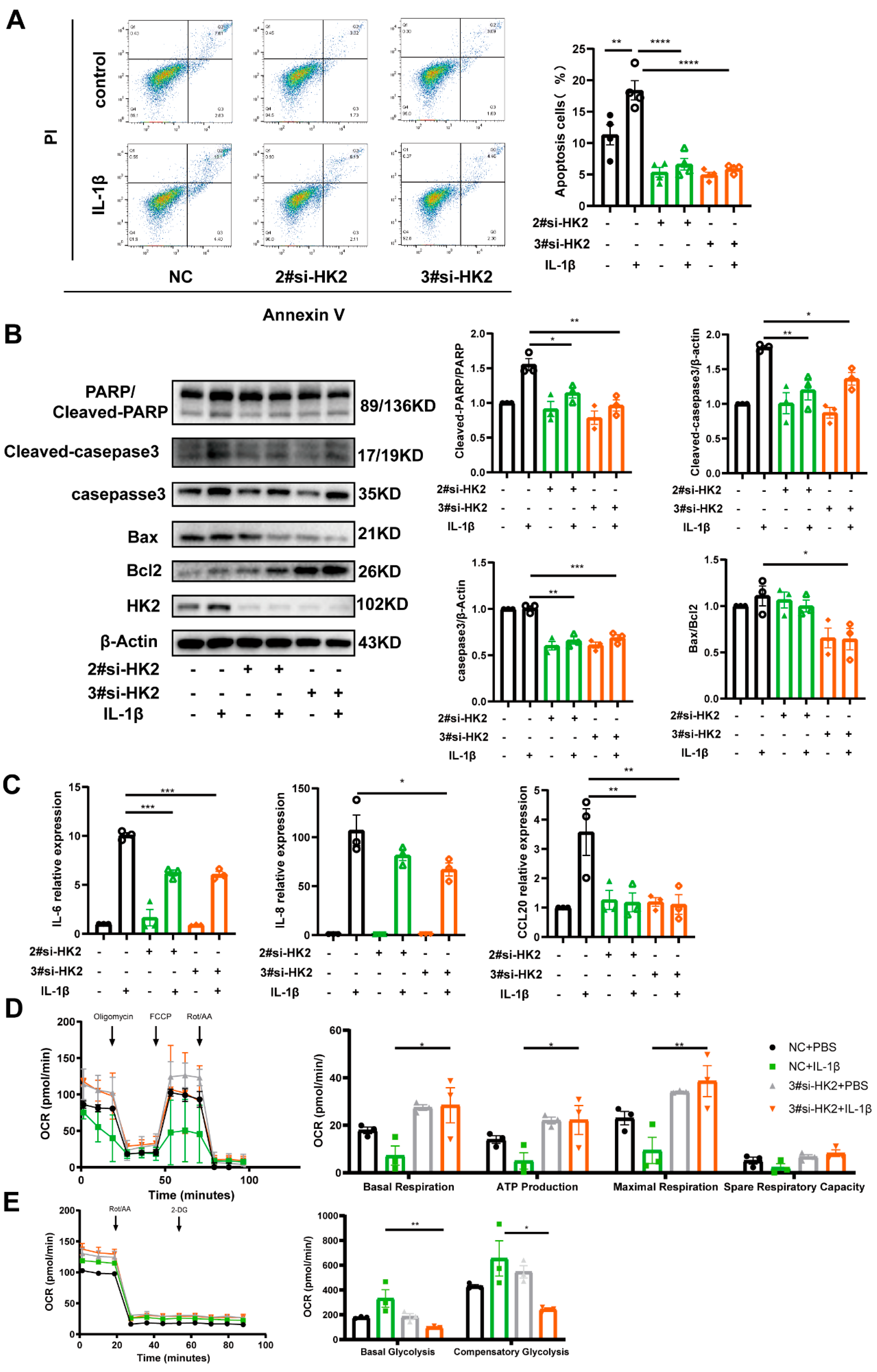
3.5. HK2 Knockdown Protects Airway Epithelial Cells Against Apoptosis and Inflammatory Cytokine Release
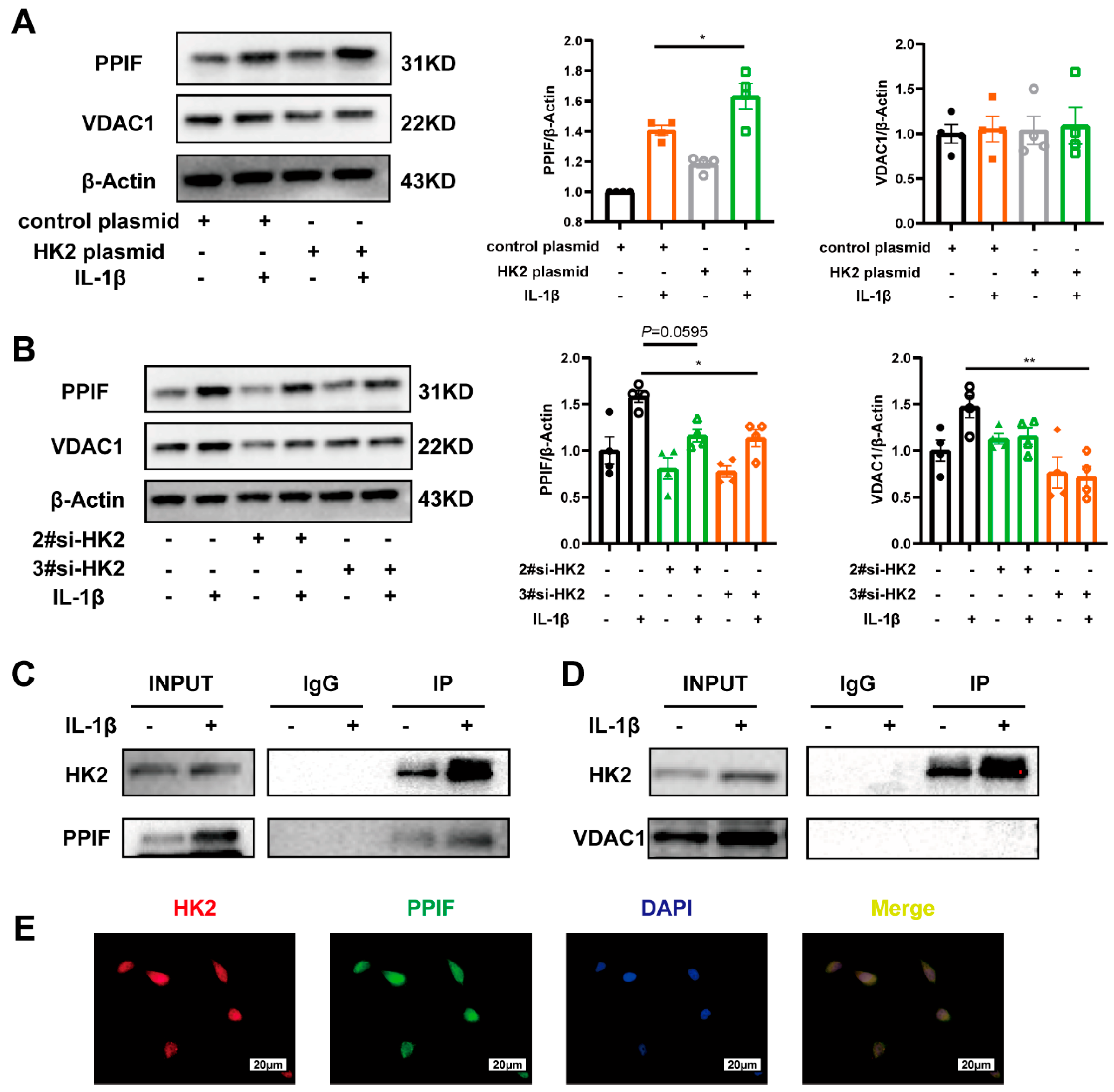
3.6. HK2 Interacts with PPIF Protecting Beas-2b Cells Against Apoptosis in Asthma
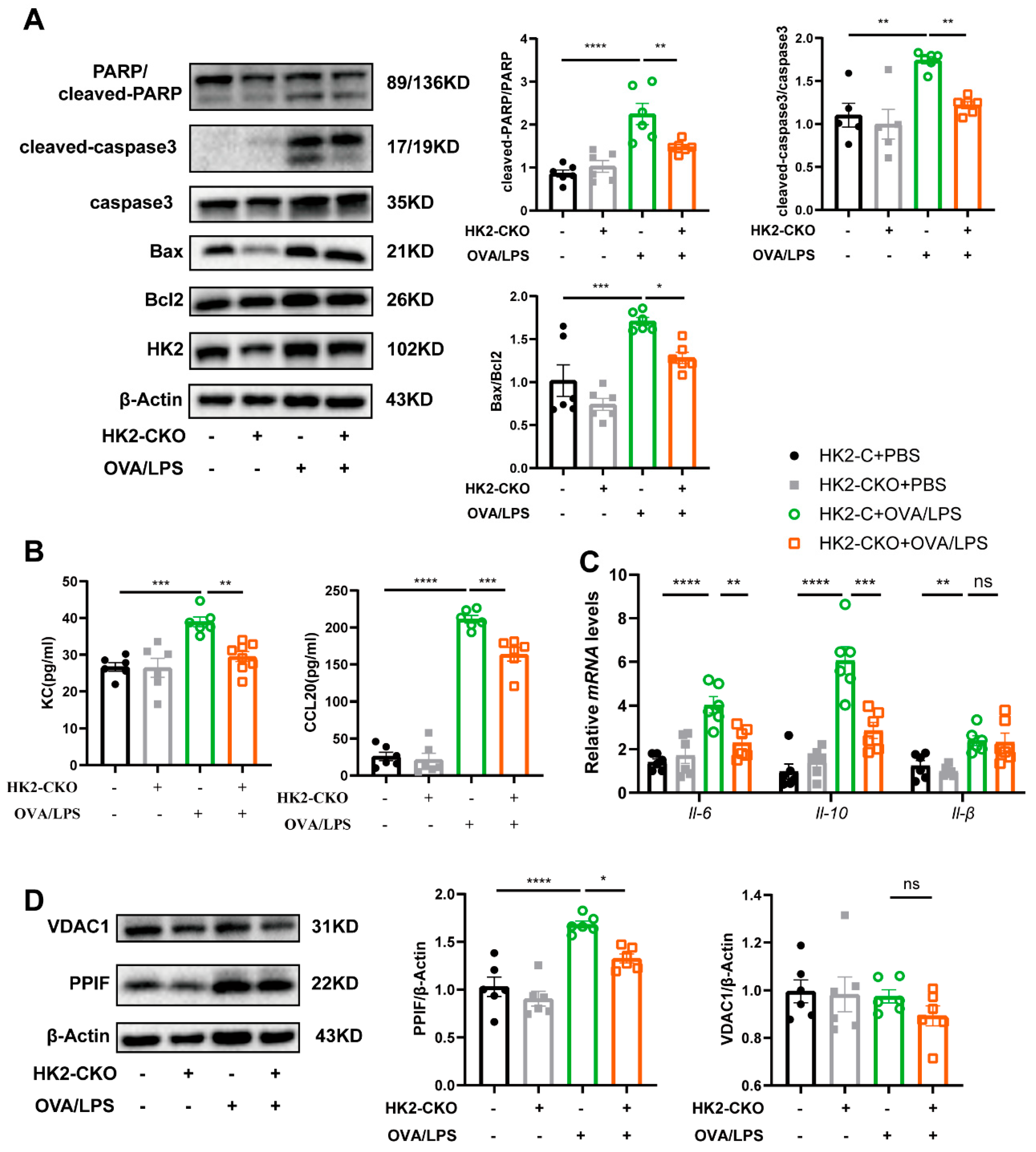
3.7. HK2 Regulates Apoptosis and Immune Response In Vivo
3.8. Inhibiting HK2 Activity by 2-DG Protected Mice from Airway Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| G6P | glucose-6-phosphate |
| OVA | ovalbumin |
| LPS | lipopolysaccharide |
| AHR | airway hyperresponsiveness |
| HK | hexokinase |
| AHR | Airway hyperresponsiveness |
| PBMC | peripheral blood mononuclear cells |
| GINA | Global Initiative for Asthma guideline |
| BALF | bronchoalveolar lavage fluid |
| HDM | house dust mite |
| IL- | Interleukin- |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| HE | hematoxylin and eosin |
| PPIF | peptidylprolyl isomerase F |
| VDAC1 | voltage-dependent anion channel 1 |
| 2-DG | 2-Deoxy-D-arabino-hexose |
| PARP1 | Poly (ADP-Ribose)-Polymerase 1 |
| Bcl-2 | B-cell lymphoma 2 protein |
| FBS | fetal bovine serum |
| Ers | respiratory system elasticity |
| Rrs | respiratory system resistance |
| Crs | respiratory system compliance |
| RNASeq | RNA sequencing |
| DEGs | differentially expressed genes |
| GOanalysis | Gene Ontology analysis |
| OCR | Oxygen consumption rate |
| FCCP | Carbonyl cyanide- 4-(trifluoromethoxy) phenylhydrazone |
| MPTP | mitochondrial permeability transition pore |
| TCA | the tricarboxylic acid cycle |
| siRNA | small-interfering RNA |
| PBMC | peripheral blood mononuclear cells |
| PBS | phosphate-buffered saline |
| HC | healthy control |
References
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. 2024. Available online: https://ginasthma.org/2024-report/ (accessed on 2 March 2025).
- Pavord, I.D.; Beasley, R.; Agusti, A.; Anderson, G.P.; Bel, E.; Brusselle, G.; Cullinan, P.; Custovic, A.; Ducharme, F.M.; Fahy, J.V.; et al. After asthma: Redefining airways diseases. Lancet 2018, 391, 350–400. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; McDonald, V.M.; Pavord, I.D.; Gibson, P.G. Asthma remission: What is it and how can it be achieved? Eur. Respir. J. 2022, 60, 2102583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin beta3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 2019, 20, 328–337. [Google Scholar] [CrossRef]
- Zheng, Y.; Lan, L.; Lu, G.; Gao, Y.-D. Patterns and trends in asthma incidence rates in main Asian and Western countries and their prediction to 2030. Chin. Med J. Pulm. Crit. Care Med. 2024, 2, 188–196. [Google Scholar] [CrossRef]
- Miethe, S.; Karsonova, A.; Karaulov, A.; Renz, H. Obesity and asthma. J. Allergy Clin. Immunol. 2020, 146, 685–693. [Google Scholar] [CrossRef]
- Reyes-Angel, J.; Kaviany, P.; Rastogi, D.; Forno, E. Obesity-related asthma in children and adolescents. Lancet Child Adolesc. Heal. 2022, 6, 713–724. [Google Scholar] [CrossRef]
- Miethe, S.; Potaczek, D.P.; Bazan-Socha, S.; Bachl, M.; Schaefer, L.; Wygrecka, M.; Garn, H. The emerging role of extracellular vesicles as communicators between adipose tissue and pathologic lungs with a special focus on asthma. Am. J. Physiol. Physiol. 2023, 324, C1119–C1125. [Google Scholar] [CrossRef]
- Kaczyńska, K.; Zając, D.; Wojciechowski, P.; Jampolska, M. Regulatory Peptides in Asthma. Int. J. Mol. Sci. 2021, 22, 13656. [Google Scholar] [CrossRef]
- Raby, K.L.; Michaeloudes, C.; Tonkin, J.; Chung, K.F.; Bhavsar, P.K. Mechanisms of airway epithelial injury and abnormal repair in asthma and COPD. Front. Immunol. 2023, 14, 1201658. [Google Scholar] [CrossRef]
- Domingo, C.; Mirapeix, R.M. From the Allergic Cascade to the Epithelium-Driven Disease: The Long Road of Bronchial Asthma. Int. J. Mol. Sci. 2023, 24, 2716. [Google Scholar] [CrossRef] [PubMed]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell–derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Miethe, S.; Schindler, V.; Alhamdan, F.; Garn, H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell Signal. 2020, 69, 109523. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Chen, Y.; Wang, Y.; Xu, Y.; Liu, T.; Mu, Q.; Huang, C. Immunometabolism in the pathogenesis of asthma. Immunology 2023, 171, 1–17. [Google Scholar] [CrossRef]
- Daley-Yates, P.; Keppler, B.; Baines, A.; Bardsley, G.; Fingleton, J. Metabolomic changes related to airway inflammation, asthma pathogenesis and systemic activity following inhaled fluticasone furoate/vilanterol: A randomized controlled trial. Respir. Res. 2022, 23, 1–17. [Google Scholar] [CrossRef]
- Li, L.; Yang, D.C.; Chen, C.-H. Metabolic reprogramming: A driver of cigarette smoke-induced inflammatory lung diseases. Free. Radic. Biol. Med. 2020, 163, 392–401. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Xu, B.; Hui, C.K.M.; Chung, K.F.; Adcock, I.M. Role of Metabolic Reprogramming in Pulmonary Innate Immunity and Its Impact on Lung Diseases. J. Innate Immun. 2019, 12, 31–46. [Google Scholar] [CrossRef]
- Barosova, R.; Baranovicova, E.; Hanusrichterova, J.; Mokra, D. Metabolomics in Animal Models of Bronchial Asthma and Its Translational Importance for Clinics. Int. J. Mol. Sci. 2023, 25, 459. [Google Scholar] [CrossRef]
- Rodríguez-Saavedra, C.; Morgado-Martínez, L.E.; Burgos-Palacios, A.; King-Díaz, B.; López-Coria, M.; Sánchez-Nieto, S. Moonlighting Proteins: The Case of the Hexokinases. Front. Mol. Biosci. 2021, 8, 701975. [Google Scholar] [CrossRef]
- Calmettes, G.; Ribalet, B.; John, S.; Korge, P.; Ping, P.; Weiss, J.N. Hexokinases and cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 107–115. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, F.; Hamm, J.; Westermann, M.; Schröder, L.; Shima, K.; Mishra, N.; Walker, A.; Sommer, N.; Klischies, K.; Prasse, D.; et al. Microbial regulation of hexokinase 2 links mitochondrial metabolism and cell death in colitis. Cell Metab. 2021, 33, 2355–2366.e8. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.J.; Fearon, U. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef]
- Bustamante, M.F.; Oliveira, P.G.; Garcia-Carbonell, R.; Croft, A.P.; Smith, J.M.; Serrano, R.L.; Sanchez-Lopez, E.; Liu, X.; Kisseleva, T.; Hay, N.; et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1636–1643. [Google Scholar] [CrossRef]
- Song, G.; Lu, Q.; Fan, H.; Zhang, X.; Ge, L.; Tian, R.; Wang, S.; Feng, T.; Pan, J.; Feng, J.; et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Steinert, E.M.; Chhiba, K.; Martinez, C.A.; Gao, P.; Perlman, H.R.; Bryce, P.; Hay, N.; Chandel, N.S. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab. 2018, 6, 10. [Google Scholar] [CrossRef]
- Williamson, M.K.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Zeidler, J.D.; Da Poian, A.T.; et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses. 2018, 10, 114. [Google Scholar] [CrossRef]
- Wang, S.; Tang, K.; Lu, Y.; Tian, Z.; Huang, Z.; Wang, M.; Zhao, J.; Xie, J. Revealing the role of glycerophospholipid metabolism in asthma through plasma lipidomics. Clin. Chim. Acta. 2021, 513, 34–42. [Google Scholar] [CrossRef]
- Alcorn, J.F.; Guala, A.S.; van der Velden, J.; McElhinney, B.; Irvin, C.G.; Davis, R.J.; Janssen-Heininger, Y.M.W. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J. Cell. Sci. 2008, 121, 1036–1045. [Google Scholar] [CrossRef]
- Lam, H.C.; Choi, A.M.; Ryter, S.W. Isolation of mouse respiratory epithelial cells and exposure to experimental cigarette smoke at air liquid interface. J. Vis. Exp. 2011, 21, 2513. [Google Scholar] [CrossRef]
- Wang, S.; Tian, Z.; Lu, Y.; Huang, Z.; Fan, Y.; Li, B.; Zheng, H.; Wu, X.; Wang, M.; Zhao, J.; et al. Sphingosine-1-Phosphate Receptor 4 Attenuates Neutrophilic Airway Inflammation in Experimental Asthma via Repressing Proinflammatory Macrophage Activation. Int. J. Biol. Sci. 2023, 19, 1597–1615. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, D.K.; Bhattacharya, A.; Mirzaei, R.; Rawji, K.S.; Ahn, Y.; Rho, J.M.; Yong, V.W. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J. Clin. Investig. 2019, 129, 3277–3292. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Aboushousha, R.; van de Wetering, C.; Chia, S.B.; Amiel, E.; Schneider, R.W.; van der Velden, J.L.J.; Lahue, K.G.; Hoagland, D.A.; Casey, D.T.; et al. IL-1/inhibitory kappaB kinase epsilon-induced glycolysis augment epithelial effector function and promote allergic airways disease. J. Allergy Clin. Immunol. 2018, 142, 435–450.e10. [Google Scholar] [CrossRef]
- Machida, K.; Ohta, Y.; Osada, H. Suppression of Apoptosis by Cyclophilin D via Stabilization of Hexokinase II Mitochondrial Binding in Cancer Cells. J. Biol. Chem. 2006, 281, 14314–14320. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef]
- Goretzki, A.; Zimmermann, J.; Rainer, H.; Lin, Y.-J.; Schülke, S. Immune Metabolism in TH2 Responses: New Opportunities to Improve Allergy Treatment—Disease-Specific Findings (Part 1). Curr. Allergy Asthma Rep. 2022, 23, 29–40. [Google Scholar] [CrossRef]
- Ye, L.; Jiang, Y.; Zhang, M. Crosstalk between glucose metabolism, lactate production and immune response modulation. Cytokine Growth Factor Rev. 2022, 68, 81–92. [Google Scholar] [CrossRef]
- Ostroukhova, M.; Goplen, N.; Karim, Z.; Michalec, L.; Guo, L.; Liang, Q.; Alam, R. The role of low-level lactate production in airway inflammation in asthma. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L300–L307. [Google Scholar] [CrossRef]
- Nowadly, C.D.; Liao, S.; Rose, J.S. Effects of Continuous Albuterol Inhalation on Serum Metabolome in Healthy Subjects: More Than Just Lactic Acid. J. Clin. Pharmacol. 2020, 61, 649–655. [Google Scholar] [CrossRef]
- Yin, X.; Choudhury, M.; Kang, J.-H.; Schaefbauer, K.J.; Jung, M.-Y.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci. Signal. 2019, 12, eaax4067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, C.; Wang, X.; He, S.; Li, Q.; Zhou, Y.; Liu, Y.; Zhang, M.; Yu, X.; Zhao, X.; et al. Lipopolysaccharide-induced proliferation and glycolysis in airway smooth muscle cells via activation of Drp1. J. Cell. Physiol. 2018, 234, 9255–9263. [Google Scholar] [CrossRef] [PubMed]
- Healey, D.C.C.; Cephus, J.Y.; Barone, S.M.; Chowdhury, N.U.; ODahunsi, D.; Madden, M.Z.; Ye, X.; Yu, X.; Olszewski, K.; Young, K.; et al. Targeting In Vivo Metabolic Vulnerabilities of Th2 and Th17 Cells Reduces Airway Inflammation. J. Immunol. 2021, 206, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Gon, Y.; Hashimoto, S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol. Int. 2018, 67, 12–17. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef]
- Li, X.; Zhao, F.; Wang, A.; Cheng, P.; Chen, H. Role and mechanisms of autophagy in lung metabolism and repair. Cell. Mol. Life Sci. 2021, 78, 5051–5068. [Google Scholar] [CrossRef]
- Li, K.; Li, M.; Li, W.; Yu, H.; Sun, X.; Zhang, Q.; Li, X.; Li, Y.; Abel, E.D.; Wu, Q.; et al. Airway epithelial regeneration requires autophagy and glucose metabolism. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis–Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef]
- Oikonomou, N.; Schuijs, M.J.; Chatzigiagkos, A.; Androulidaki, A.; Aidinis, V.; Hammad, H.; Lambrecht, B.N.; Pasparakis, M. Airway epithelial cell necroptosis contributes to asthma exacerbation in a mouse model of house dust mite-induced allergic inflammation. Mucosal Immunol. 2021, 14, 1160–1171. [Google Scholar] [CrossRef]
- Persson, C. Airways exudation of plasma macromolecules: Innate defense, epithelial regeneration, and asthma. J. Allergy Clin. Immunol. 2018, 143, 1271–1286. [Google Scholar] [CrossRef]
- Thiriou, D.; Morianos, I.; Xanthou, G.; Samitas, K. Innate immunity as the orchestrator of allergic airway inflammation and resolution in asthma. Int. Immunopharmacol. 2017, 48, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Dicato, M.; Diederich, M. Modulatory roles of glycolytic enzymes in cell death. Biochem. Pharmacol. 2014, 92, 22–30. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Weiss, J.N.; Ribalet, B.; Rodrigues-Lima, F. Subcellular Localization of Hexokinases I and II Directs the Metabolic Fate of Glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Z.; Zheng, H.; Fan, Y.; Li, B.; Huang, Z.; Wang, M.; Zhang, J.; Zhao, J.; Wang, S.; Xie, J. Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F. Cells 2025, 14, 1004. https://doi.org/10.3390/cells14131004
Tian Z, Zheng H, Fan Y, Li B, Huang Z, Wang M, Zhang J, Zhao J, Wang S, Xie J. Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F. Cells. 2025; 14(13):1004. https://doi.org/10.3390/cells14131004
Chicago/Turabian StyleTian, Zhen, Hongyan Zheng, Yan Fan, Boyu Li, Zhenli Huang, Meijia Wang, Jixian Zhang, Jianping Zhao, Shanshan Wang, and Jungang Xie. 2025. "Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F" Cells 14, no. 13: 1004. https://doi.org/10.3390/cells14131004
APA StyleTian, Z., Zheng, H., Fan, Y., Li, B., Huang, Z., Wang, M., Zhang, J., Zhao, J., Wang, S., & Xie, J. (2025). Upregulated Hexokinase-2 in Airway Epithelium Regulates Apoptosis and Drives Inflammation in Asthma via Peptidylprolyl Isomerase F. Cells, 14(13), 1004. https://doi.org/10.3390/cells14131004