
Role of Nitric Oxide in Cardioprotection by Poloxamer 188



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
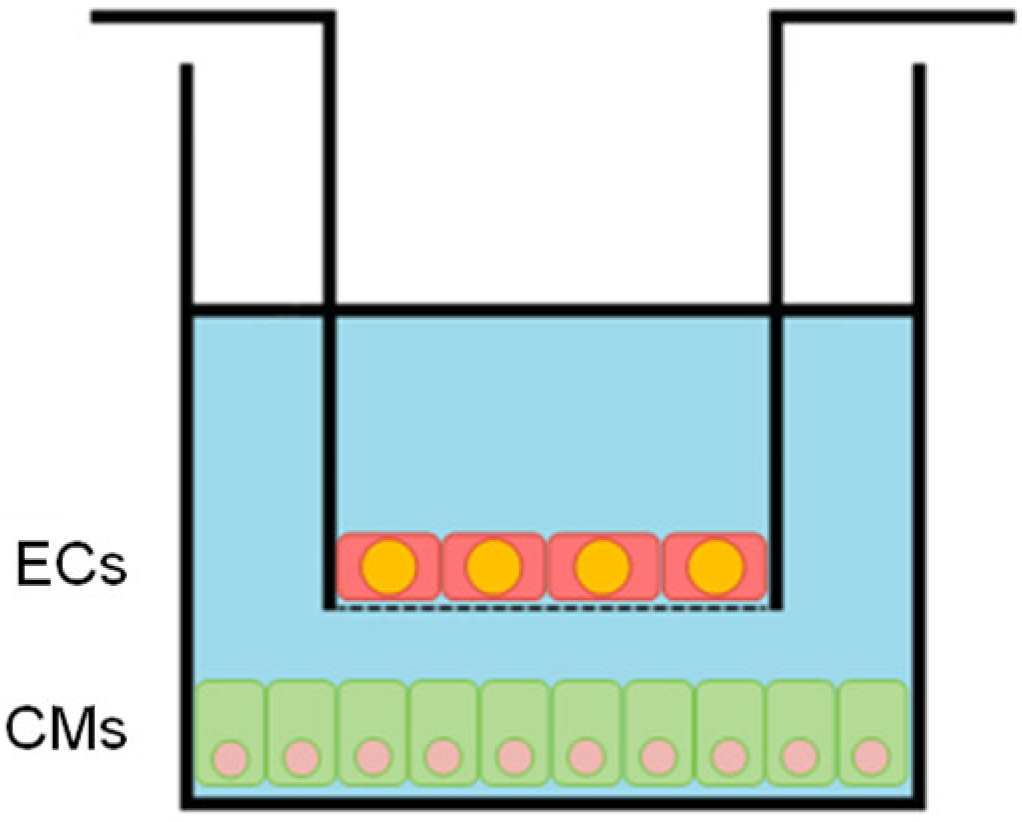
2.1. Cell Culture
2.2. In Vitro HR Injury
2.3. LDH Cytotoxicity Assay
2.4. Fluo-4 Direct Calcium Assay
2.5. Treatment with P188 and LNAME
2.6. Statistics
3. Results
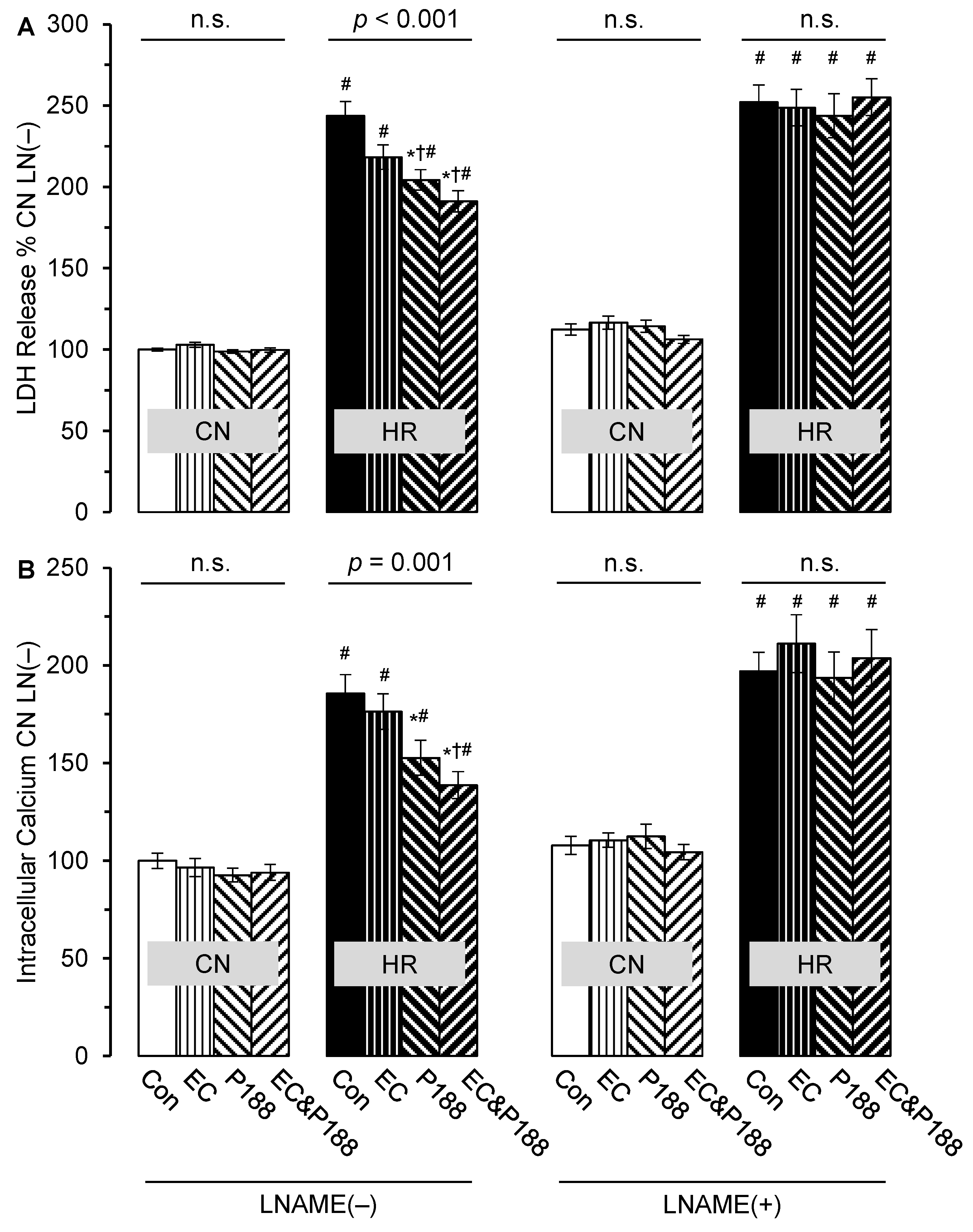
3.1. LDH Release
3.2. Intracellular Calcium
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CM(s) | Cardiomyocyte(s) |
| CN | Control normoxia |
| EC(s) | Endothelial cell(s) |
| HR | Hypoxia reoxygenation |
| IR | Ischemia reperfusion |
| LDH | Lactate dehydrogenase |
| LNAME | N(ω)-nitro-L-arginine methyl ester |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| P | Poloxamer |
| PEO | Poly-propylene oxide |
| PPO | Poly-ethylene oxide |
| SGF | Serum- and glucose-free |
References
- Lefer, A.M.; Tsao, P.S.; Lefer, D.J.; Ma, X.L. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J. 1991, 5, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K. Endothelial dysfunction and reactive oxygen species production in ischemia/reperfusion and nitrate tolerance. Gen. Physiol. Biophys. 2004, 23, 265–295. [Google Scholar] [PubMed]
- Perry, M.O.; Fantini, G. Ischemia-reperfusion and cell membrane dysfunction. Microcirc. Endothel. Lymphat. 1989, 5, 241–258. [Google Scholar]
- Cipolla, M.J.; Chan, S.L.; Sweet, J.; Tavares, M.J.; Gokina, N.; Brayden, J.E. Postischemic reperfusion causes smooth muscle calcium sensitization and vasoconstriction of parenchymal arterioles. Stroke 2014, 45, 2425–2430. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef]
- Wang, R.; Wang, M.; He, S.; Sun, G.; Sun, X. Targeting Calcium Homeostasis in Myocardial Ischemia/Reperfusion Injury: An Overview of Regulatory Mechanisms and Therapeutic Reagents. Front. Pharmacol. 2020, 11, 872. [Google Scholar] [CrossRef]
- Jaeschke, H.; Lemasters, J.J. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 2003, 125, 1246–1257. [Google Scholar] [CrossRef]
- Eefting, F.; Rensing, B.; Wigman, J.; Pannekoek, W.J.; Liu, W.M.; Cramer, M.J.; Lips, D.J.; Doevendans, P.A. Role of apoptosis in reperfusion injury. Cardiovasc. Res. 2004, 61, 414–426. [Google Scholar] [CrossRef]
- Gottlieb, R.A. Cell death pathways in acute ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 233–238. [Google Scholar] [CrossRef]
- Logue, S.E.; Gustafsson, A.B.; Samali, A.; Gottlieb, R.A. Ischemia/reperfusion injury at the intersection with cell death. J. Mol. Cell. Cardiol. 2005, 38, 21–33. [Google Scholar] [CrossRef]
- Moloughney, J.G.; Weisleder, N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat. Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Bartos, J.A.; Matsuura, T.R.; Sarraf, M.; Youngquist, S.T.; McKnite, S.H.; Rees, J.N.; Sloper, D.T.; Bates, F.S.; Segal, N.; Debaty, G.; et al. Bundled postconditioning therapies improve hemodynamics and neurologic recovery after 17 min of untreated cardiac arrest. Resuscitation 2015, 87, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bartos, J.A.; Matsuura, T.R.; Tsangaris, A.; Olson, M.D.; McKnite, S.H.; Rees, J.N.; Haman, K.; Chandra Shekar, K.; Riess, M.L.; Bates, F.S.; et al. Intracoronary Poloxamer 188 Prevents Reperfusion Injury in a Porcine Model of ST-Segment Elevation Myocardial Infarction. JACC. Basic Transl. Sci. 2016, 1, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock polymers: Panacea or Pandora’s box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef]
- Lee, R.C.; River, L.P.; Pan, F.S.; Ji, L.; Wollmann, R.L. Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4524–4528. [Google Scholar] [CrossRef]
- Lee, R.C.; Myerov, A.; Maloney, C.P. Promising therapy for cell membrane damage. Ann. N. Y. Acad. Sci. 1994, 720, 239–245. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Poloxamer 188 volumetrically decreases neuronal loss in the rat in a time-dependent manner. Neurosurgery 2004, 55, 943–948, discussion 948-949. [Google Scholar] [CrossRef]
- Forman, M.B.; Ingram, D.A.; Murray, J.J. Role of perfluorochemical emulsions in the treatment of myocardial reperfusion injury. Am. Heart J. 1992, 124, 1347–1357. [Google Scholar] [CrossRef]
- Martindale, J.J.; Metzger, J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J. Mol. Cell. Cardiol. 2014, 67, 26–37. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, L.; He, F.; Chen, J.; Zhao, M.; Li, S.; Wu, H.; Liu, Y.; Zhang, Y.; Ping, Q.; et al. Surfactant Assisted Rapid-Release Liposomal Strategies Enhance the Antitumor Efficiency of Bufalin Derivative and Reduce Cardiotoxicity. Int. J. Nanomed. 2021, 16, 3581–3598. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, D.; Ito, Y.; Liu, X.; Zhang, J.; Wu, C. An ocular drug delivery system containing zinc diethyldithiocarbamate and HPbetaCD inclusion complex--corneal permeability, anti-cataract effects and mechanism studies. J. Pharm. Pharmacol. 2004, 56, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F.; Guerron, A.D.; Yu, Q.; Sali, A.; van der Meulen, J.H.; Hoffman, E.P.; Nagaraju, K. Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc. Disord. 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Salzman, M.M.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Protects Isolated Adult Mouse Cardiomyocytes from Reoxygenation Injury. Pharmacol. Res. Perspect. 2020, 8, e00639. [Google Scholar] [CrossRef]
- Li, Z.; Gupta, M.K.; Barajas, M.B.; Oyama, T.; Duvall, C.L.; Riess, M.L. Newly Developed Di-Block Copolymer-Based Cell Membrane Stabilizers Protect Mouse Coronary Artery Endothelial Cells against Hypoxia/Reoxygenation Injury. Cells 2023, 12, 1394. [Google Scholar] [CrossRef]
- Chen, G.; Douglas, H.F.; Li, Z.; Cleveland, W.J.; Balzer, C.; Yannopoulos, D.; Chen, I.Y.; Obal, D.; Riess, M.L. Cardioprotection by poloxamer 188 is mediated through increased endothelial nitric oxide production. Sci. Rep. 2025, 15, 15170. [Google Scholar] [CrossRef]
- Maskarinec, S.A.; Hannig, J.; Lee, R.C.; Lee, K.Y. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys. J. 2002, 82, 1453–1459. [Google Scholar] [CrossRef]
- Serbest, G.; Horwitz, J.; Jost, M.; Barbee, K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006, 20, 308–310. [Google Scholar] [CrossRef]
- Leucker, T.M.; Bienengraeber, M.; Muravyeva, M.; Baotic, I.; Weihrauch, D.; Brzezinska, A.K.; Warltier, D.C.; Kersten, J.R.; Pratt, P.F., Jr. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J. Mol. Cell. Cardiol. 2011, 51, 803–811. [Google Scholar] [CrossRef]
- Leucker, T.M.; Ge, Z.D.; Procknow, J.; Liu, Y.; Shi, Y.; Bienengraeber, M.; Warltier, D.C.; Kersten, J.R. Impairment of endothelial-myocardial interaction increases the susceptibility of cardiomyocytes to ischemia/reperfusion injury. PLoS ONE 2013, 8, e70088. [Google Scholar] [CrossRef]
- Li, Z.; Hampton, M.J.W.; Barajas, M.B.; Riess, M.L. Development of a Cell Co-Culture Model to Mimic Cardiac Ischemia/Reperfusion In Vitro. J. Vis. Exp. JoVE 2021. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Haman, K.J.; Kim, M.; Zhang, W.; Lowe, D.A.; Sham, Y.Y.; Lodge, T.P.; Hackel, B.J.; Bates, F.S.; Metzger, J.M. Chemical End Group Modified Diblock Copolymers Elucidate Anchor and Chain Mechanism of Membrane Stabilization. Mol. Pharm. 2017, 14, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Flather, M.; Gent, M.; Roberts, R.; Hall, B.; OConnor, M.; Gibbons, R.; Burns, R.; Anderson, J.; Bristow, D.; et al. Effects of RheothRx on mortality, morbidity, left ventricular function, and infarct size in patients with acute myocardial infarction. Collaborative Organization for RheothRx Evaluation (CORE). Circulation 1997, 96, 192–201. [Google Scholar]
- Sandor, B.; Marin, M.; Lapoumeroulie, C.; Rabai, M.; Lefevre, S.D.; Lemonne, N.; El Nemer, W.; Mozar, A.; Francais, O.; Le Pioufle, B.; et al. Effects of Poloxamer 188 on red blood cell membrane properties in sickle cell anaemia. Br. J. Haematol. 2016, 173, 145–149. [Google Scholar] [CrossRef]
- Kim, M.; Haman, K.J.; Houang, E.M.; Zhang, W.; Yannopoulos, D.; Metzger, J.M.; Bates, F.S.; Hackel, B.J. PEO-PPO diblock copolymers protect myoblasts from hypo-osmotic stress in vitro dependent on copolymer size, composition, and architecture. Biomacromolecules 2017, 18, 2090–2101. [Google Scholar] [CrossRef]
- Wu, R.; Koduri, R.; Cho, M.; Alatrash, N.; Nomellini, V. Effects of poloxamer 188 on traumatic brain injury. Brain Behav. Immun.-Health 2024, 38, 100762. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2020, 598, 2923–2939. [Google Scholar] [CrossRef]
- Totzeck, M.; Hendgen-Cotta, U.B.; Rassaf, T. Nitrite-Nitric Oxide Signaling and Cardioprotection. Adv. Exp. Med. Biol. 2017, 982, 335–346. [Google Scholar] [CrossRef]
- Luiking, Y.C.; Engelen, M.P.; Deutz, N.E. Regulation of nitric oxide production in health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 97–104. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Barajas, M.B.; Oyama, T.; Riess, M.L. Role of Nitric Oxide in Cardioprotection by Poloxamer 188. Cells 2025, 14, 1001. https://doi.org/10.3390/cells14131001
Li Z, Barajas MB, Oyama T, Riess ML. Role of Nitric Oxide in Cardioprotection by Poloxamer 188. Cells. 2025; 14(13):1001. https://doi.org/10.3390/cells14131001
Chicago/Turabian StyleLi, Zhu, Matthew B. Barajas, Takuro Oyama, and Matthias L. Riess. 2025. "Role of Nitric Oxide in Cardioprotection by Poloxamer 188" Cells 14, no. 13: 1001. https://doi.org/10.3390/cells14131001
APA StyleLi, Z., Barajas, M. B., Oyama, T., & Riess, M. L. (2025). Role of Nitric Oxide in Cardioprotection by Poloxamer 188. Cells, 14(13), 1001. https://doi.org/10.3390/cells14131001