
Spatial Transcriptomics in Thyroid Cancer: Applications, Limitations, and Future Perspectives


Abstract
1. Introduction
2. Overview of ST
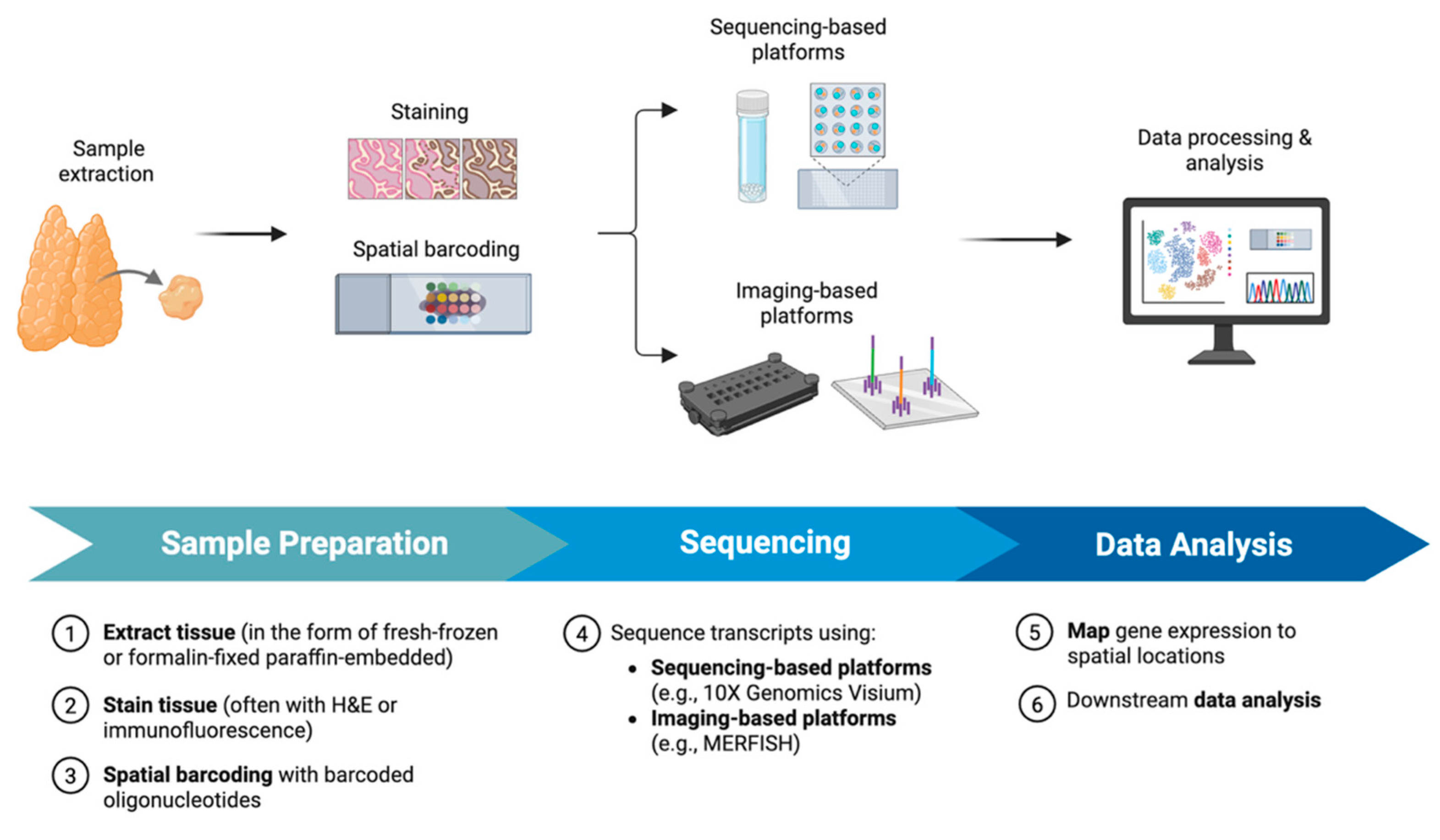
2.1. Workflow and Methodology of ST
2.2. ST Platforms
3. Spatial Heterogeneity of the Tumor Microenvironment (TME)
3.1. Cell Types and Spatial Distribution
3.1.1. Immune Cells
3.1.2. Cancer-Associated Fibroblasts (CAFs)
3.1.3. Colocalization
3.1.4. Mixed Tumors
3.2. Gene Expression Patterns and Pathway Enrichment
3.3. Subpopulation Diversity
3.4. Tumor Leading Edge
4. Tumor Evolution
4.1. Evolutionary Routes
4.2. Evolution of Subpopulations
4.3. Invasion and Metastasis
5. Cell–Cell Interactions
5.1. Identification of Signaling Pathways
5.2. Interaction Between Immune Cells and Tumor Cells
6. Limitations and Future Directions
6.1. Technical Limitations
6.2. Sample Preparation
6.3. Data Analysis and Interpretation
6.4. Clinical Applications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| AFC | Atypical thyroid follicular cells |
| ATC | Anaplastic thyroid carcinoma |
| CAF | Cancer-associated fibroblast |
| DEG | Differentially expressed gene |
| DSP | Digital Spatial Profiling |
| EMT | Epithelial–mesenchymal transition |
| FC | Follicular cells |
| FFPE | Formalin-fixed, paraffin-embedded |
| FTC | Follicular thyroid carcinoma |
| H&E | Hematoxylin and eosin |
| iCAF | Inflammatory CAF |
| IHC | Immunohistochemistry |
| LPTC | Locally advanced papillary thyroid carcinoma |
| MAP | Molecular Aggression and Prediction |
| MERFISH | Multiplexed error-robust fluorescence in situ hybridization |
| myCAF | Myofibroblastic CAF |
| PDTC | Poorly differentiated thyroid carcinoma |
| PT | Para-tumor |
| PTC | Papillary thyroid carcinoma |
| RNA-seq | RNA sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| SeqFISH | Sequential fluorescence in situ hybridization |
| SMI | Spatial Molecular Imager |
| ST | Spatial transcriptomics |
| TCGA | The Cancer Genome Atlas |
| THCA | Thyroid cancer |
| TME | Tumor microenvironment |
References
- Siegel, R.; Miller, K.; Wagle, N.; Jemal, A. Cancer statistics 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- LeClair, K.; Bell, K.J.L.; Furuya-Kanamori, L.; Doi, S.A.; Francis, D.O.; Davies, L. Evaluation of gender inequity in thyroid cancer diagnosis: Differences by sex in US thyroid cancer incidence compared with a meta-analysis of subclinical thyroid cancer rates at autopsy. JAMA Intern. Med. 2021, 181, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Hundahl, S.A.; Fleming, I.D.; Fremgen, A.M.; Menck, H.R. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995. Cancer 1998, 83, 2638–2648. [Google Scholar] [CrossRef]
- Kebebew, E.; Greenspan, F.S.; Clark, O.H.; Woeber, K.A.; McMillan, A. Anaplastic thyroid carcinoma: Treatment outcome and prognostic factors. Cancer 2005, 103, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Nixon, I.J.; Ganly, I.; Patel, S.G.; Palmer, F.L.; Whitcher, M.M.; Tuttle, R.M.; Shaha, A.; Shah, J.P. Thyroid lobectomy for treatment of well differentiated intrathyroid malignancy. Surgery 2012, 151, 571–579. [Google Scholar] [CrossRef]
- Seidlin, S.M.; Marinelli, L.D.; Oshry, E. Radioactive iodine therapy: Effect on functioning metastases of adenocarcinoma of the thyroid. CA Cancer J. Clin. 1990, 40, 299–317. [Google Scholar] [CrossRef]
- Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Sherman, S.I.; Tuttle, R.M.; et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef]
- Pešorda, M.; Kusačić Kuna, S.; Huić, D.; Herceg, D.; Despot, M.; Samardžić, T.; Gnjidić, M.; Belev, B. Kinase inhibitors in the treatment of thyroid cancer: Institutional experience. Acta Clin. Croat. 2020, 59 (Suppl. S1), 73–80. [Google Scholar] [CrossRef]
- Iyer, P.C.; Dadu, R.; Gule-Monroe, M.; Busaidy, N.L.; Ferrarotto, R.; Habra, M.A.; Zafereo, M.; Williams, M.D.; Gunn, G.B.; Grosu, H.; et al. Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J. Immunother. Cancer 2018, 6, 68. [Google Scholar] [CrossRef]
- Pu, W.; Shi, X.; Yu, P.; Zhang, M.; Liu, Z.; Tan, Y.; Li, J.; Xie, L.; Yang, L.; Ma, J. Single-cell transcriptomic analysis of the tumor ecosystems underlying initiation and progression of papillary thyroid carcinoma. Nat. Commun. 2021, 12, 6058. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.; Cen, X.; Xu, J.; Zhao, S.; Wang, B.; Zhang, W.; Qiu, M. Integrated analysis of fibroblasts molecular features in papillary thyroid cancer combining single-cell and bulk RNA sequencing technology. Front. Endocrinol. 2022, 13, 1019072. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Cong, S.; Wang, Y.; Ji, Q.; Liu, W.; Qu, N. Analysis of the key ligand receptor CADM1_CADM1 in the regulation of thyroid cancer based on scRNA-seq and bulk RNA-seq data. Front. Endocrinol. 2022, 13, 969914. [Google Scholar] [CrossRef]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clément, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Fernandéz Navarro, J.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Affinito, O.; Orlandella, F.M.; Luciano, N.; Salvatore, M.; Salvatore, G.; Franzese, M. Evolution of intra-tumoral heterogeneity across different pathological stages in papillary thyroid carcinoma. Cancer Cell Int. 2022, 22, 263. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- 10× Genomics. Visium Spatial Assays. 14 May 2025. Available online: https://www.10xgenomics.com/platforms/visium/product-family (accessed on 14 May 2025).
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zheng, G.; Chen, S.; Ma, W.; Wang, Q.; Sun, L.; Zhang, C.; Chen, G.; Zhang, S.; Chen, S. Spatial and single-cell transcriptomics unraveled spatial evolution of papillary thyroid cancer. Adv. Sci. 2025, 12, e2404491. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Liu, Q.-Z.; Huang, R.-R.; Jiang, Y.-H.; Bian, Z.-H.; Li, S.-J.; Li, L.; Shen, F.; Tsuneyama, K.; Zhang, Q.-L.; et al. Spatial transcriptomics reveals prognosis-associated cellular heterogeneity in the papillary thyroid carcinoma microenvironment. Clin. Transl. Med. 2024, 14, e1594. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ji, X.; Zhang, Y.; Yang, F.; Su, H.; Zhang, H.; Li, Z.; Zhang, W.; Sun, W. Interactions between LAMP3+ dendritic cells and T-cell subpopulations promote immune evasion in papillary thyroid carcinoma. J. Immunother. Cancer 2024, 12, e008983. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Han, P.-Z.; Ye, W.-D.; Yu, P.-C.; Tan, L.-C.; Shi, X.; Chen, X.-F.; He, C.; Hu, J.-Q.; Wei, W.-J.; Lu, Z.-W.; et al. A distinct tumor microenvironment makes anaplastic thyroid cancer more lethal but immunotherapy sensitive than papillary thyroid cancer. JCI Insight 2024, 9, e173712. [Google Scholar] [CrossRef]
- Luo, H.; Xia, X.; Kim, G.D.; Liu, Y.; Xue, Z.; Zhang, L.; Shu, Y.; Yang, T.; Chen, Y.; Zhang, S.; et al. Characterizing dedifferentiation of thyroid cancer by integrated analysis. Sci. Adv. 2021, 7, eabf3657. [Google Scholar] [CrossRef]
- Lu, L.; Wang, J.R.; Henderson, Y.C.; Bai, S.; Yang, J.; Hu, M.; Shiau, C.-K.; Pan, T.; Yan, Y.; Tran, T.M.; et al. Anaplastic transformation in thyroid cancer revealed by single-cell transcriptomics. J. Clin. Investig. 2023, 133, e169653. [Google Scholar] [CrossRef]
- Loberg, M.A.; Xu, G.J.; Chen, S.-C.; Chen, H.-C.; Wahoski, C.C.; Caroland, K.P.; Tigue, M.L.; Hartmann, H.A.; Gallant, J.-N.; Phifer, C.J.; et al. An integrated single-cell and spatial transcriptomic atlas of thyroid cancer progression identifies prognostic fibroblast subpopulations. bioRxiv 2025. [Google Scholar] [CrossRef]
- Tourneur, A.; Vitória, J.R.; Saiselet, M.; Craciun, L.; Larsimont, D.; Lefort, A.; Libert, F.; Maenhaut, C.; Costagliola, S.; Tarabichi, M.; et al. Single nuclei and spatial transcriptomes suggest a stratification of papillary and anaplastic thyroid cancer cells. bioRxiv 2024. [Google Scholar] [CrossRef]
- Xu, G.J.; Loberg, M.A.; Gallant, J.-N.; Sheng, Q.; Chen, S.-C.; Lehmann, B.D.; Shaddy, S.M.; Tigue, M.L.; Phifer, C.J.; Wang, L.; et al. Molecular signature incorporating the immune microenvironment enhances thyroid cancer outcome prediction. Cell Genom. 2023, 3, 100409. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, R.; Yan, F.; Lin, Y.; Wang, H.; Xiong, H. Analysis of thyroid carcinoma composition and spatial architecture in the progression of dedifferentiation, lymphatic metastasis, and gastric metastasis. J. Transl. Med. 2025, 23, 213. [Google Scholar] [CrossRef]
- Liao, T.; Zeng, Y.; Xu, W.; Shi, X.; Shen, C.; Du, Y.; Zhang, M.; Zhang, Y.; Li, L.; Ding, P.; et al. A spatially resolved transcriptome landscape during thyroid cancer progression. Cell Rep. Med. 2025, 6, 102043. [Google Scholar] [CrossRef]
- Haq, F.; Bychkov, A.; Mete, O.; Jeon, S.; Jung, C.K. Identification of specific biomarkers for anaplastic thyroid carcinoma through spatial transcriptomic and immunohistochemical profiling. Endocr. Pathol. 2025, 36, 14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Nojima, S.; Tahara, S.; Motooka, D.; Kohara, M.; Okuzaki, D.; Hirokawa, M.; Morii, E. Identification of invasive subpopulations using spatial transcriptome analysis in thyroid follicular tumors. J. Pathol. Transl. Med. 2024, 58, 22–28. [Google Scholar] [CrossRef]
- Condello, V.; Paulsson, J.O.; Zedenius, J.; Näsman, A.; Juhlin, C.C. Spatial transcriptomics in a case of follicular thyroid carcinoma reveals clone-specific dysregulation of genes regulating extracellular matrix in the invading front. Endocr. Pathol. 2024, 35, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Qu, N.; Zhu, R.; Hu, J.; Han, P.; Wu, J.; Tan, L.; Gan, H.; He, C.; Fang, C.; et al. TERT accelerates BRAF mutant–induced thyroid cancer dedifferentiation and progression by regulating ribosome biogenesis. Sci. Adv. 2023, 9, eadg7125. [Google Scholar] [CrossRef]
- Xiao, G.; Xie, R.; Gu, J.; Huang, Y.; Ding, M.; Shen, D.; Yan, J.; Yuan, J.; Yang, Q.; He, W.; et al. Single-cell RNA-sequencing and spatial transcriptomic analysis reveal a distinct population of APOE−; cells yielding pathological lymph node metastasis in papillary thyroid cancer. Clin. Transl. Med. 2025, 15, e70172. [Google Scholar] [CrossRef]
- Lin, C.-M.; Yu, C.-F.; Huang, H.-Y.; Chen, F.-H.; Hong, J.-H.; Chiang, C.-S. Distinct tumor microenvironment at tumor edge as a result of astrocyte activation is associated with therapeutic resistance for brain tumor. Front. Oncol. 2019, 9, 307. [Google Scholar] [CrossRef]
- Kim, J.; Parrish, A.; Kurokawa, M.; Matsuura, K.; Freel, C.; Andersen, J.; Johnson, C.; Kornbluth, S. Rsk-mediated phosphorylation and 14-3-3ε binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO J. 2012, 2012, 1279–1292. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 2000, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tran, V.; Vemuri, V.N.P.; Byrne, A.; Borja, M.; Kim, Y.J.; Agarwal, S.; Wang, R.; Awayan, K.; Murti, A.; et al. Concordance of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing. Life Sci. Alliance 2022, 6, e202201701. [Google Scholar] [CrossRef]
- Shah, S.; Lubeck, E.; Zhou, W.; Cai, L. seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron 2017, 94, 752–758.e1. [Google Scholar] [CrossRef] [PubMed]
- Bergenstråhle, L.; He, B.; Bergenstråhle, J.; Abalo, X.; Mirzazadeh, R.; Thrane, K.; Ji, A.L.; Andersson, A.; Larsson, L.; Stakenborg, N.; et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 2022, 40, 476–479. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef]
- Biancalani, T.; Scalia, G.; Buffoni, L.; Avasthi, R.; Lu, Z.; Sanger, A.; Tokcan, N.; Vanderburg, C.R.; Segerstolpe, Å.; Zhang, M.; et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 2021, 18, 1352–1362. [Google Scholar] [CrossRef]
- Canene-Adams, K. Preparation of Formalin-fixed Paraffin-embedded Tissue for Immunohistochemistry. Methods Enzymol. 2013, 533, 225–233. [Google Scholar] [CrossRef]
- Reis, P.P.; Waldron, L.; Goswami, R.S.; Xu, W.; Xuan, Y.; Perez-Ordonez, B.; Gullane, P.; Irish, J.; Jurisica, I.; Kamel-Reid, S. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol. 2011, 11, 46. [Google Scholar] [CrossRef]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. 10× Development Teams, & Taylor, S.E.B. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef]
- Badve, S.S.; Gökmen-Polar, Y. Protein profiling of breast cancer for treatment decision-making. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, e351207. [Google Scholar] [CrossRef] [PubMed]
- Góes da Costa, G.; Bombardelli Gomig, T.H.; Kaviski, R.; Sousa, K.S.; Kukolj, C.; de Lima, R.S.; Urban, C.A.; Cavalli, I.J.; Ribeiro, E.M.S.F. Comparative proteomics of tumor and paired normal breast tissue highlights potential biomarkers in breast cancer. Cancer Genom. Proteom. 2015, 12, 251–261. [Google Scholar]
- Ospina, O.; Manjarres-Betancur, R.; Gonzalez-Calderon, G.; Soupir, A.C.; Smalley, I.; Tsai, K.Y.; Markowitz, J.; Khaled, M.L.; Vallebuona, E.; Berglund, A.E.; et al. spatialGE Is a User-Friendly Web Application That Facilitates Spatial Transcriptomics Data Analysis. Cancer Res. 2025, 85, 848–858. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene(s)/Pathway(s) | Observed in | Implication | Reference |
|---|---|---|---|
| TFF3, SLC34A2 | PT, PTC | Low involvement in cancer pathways | Liao et al., 2025 [34] |
| CXCL14, SAA1 | LPTC, ATC | Biomarkers of ATC progression | Liao et al., 2025 [34] |
| COL7A1, LAMC2, SPHK1, SRPX2 | ATC | Haq et al., 2025 [35] | |
| TTF1, TG, PAX8 | ATC, PTC | Wang et al., 2025 [33] | |
| CD74 | Invasive areas of FTC | Biomarkers of FTC invasion | Suzuki et al., 2025 [36] |
| DPYSL3, POSTN, TERT, EMT-related genes | Core and invasive front of FTC | Condello et al., 2024 [37] | |
| Negative regulation of smooth muscle cell migration; collagen-containing extracellular matrix; collagen fibril organization | Vascular invasive fronts of FTC | Mechanisms for tumor invasion, survival, and/or drug resistance at the vascular area | Condello et al., 2024 [37] |
| Extracellular matrix assembly; extracellular matrix organization | Capsular invasive fronts of FTC | Mechanisms for tumor invasion, survival, and/or drug resistance at the capsular area | Condello et al., 2024 [37] |
| rRNA processing; translation | Thyrocytes in the final dedifferentiation stage | Mechanisms thyrocytes are involved in as the tumor progresses; therapeutic targets | Yu et al., 2023 [38] |
| Lipid metabolism (ferroptosis) | Lymph node metastasis of PTC | Zheng et al., 2024 [22] | |
| SERPINE1; PLAUR | ATC | Biomarker for ATC; targetable axis for ATC treatment | Liao et al., 2025 [34] |
| FN1-SDC4; FN1-ITGA3; LAMB3-ITGA2 | PTC | Targetable axes for PTC treatment | Yan et al., 2024 [23] |
| TGFβ; TGFBR1; TGFBR2 | PTC | Crosstalk between immune cells and tumor cells; targets for immunotherapy | Yan et al., 2024 [23] |
| CD6- and F11R-driven signaling pathways | PTC | Xiao et al., 2025 [39] |
| Challenge | Description | Future Direction |
|---|---|---|
| Low resolution in sequencing-based platforms (e.g., 10× Genomics Visium) | Cannot reach true single-cell or subcellular resolution | Develop higher-resolution or single-cell ST platforms (e.g., Stereo-seq, CosMx); build more advanced AI models and algorithms; combine imaging and sequencing-based approaches |
| Limited gene detection in imaging-based platforms (e.g., MERFISH, seqFISH) | Only preselected genes are detected | |
| Shallow transcriptome coverage | Capture only a fraction of expressed genes | |
| Sample preparation | Fresh frozen tissues are often required; FFPE compatibility and sensitivity are limited in some platforms | Improve protocols for FFPE compatibility to expand clinical use |
| Technical complexity | Technically demanding protocols | Standardize workflows and develop user-friendly commercial kits |
| Data integration | ST data are complex and hard to integrate with scRNA-seq or proteomics | Advance computational tools for multi-omics integration |
| High cost | Need for high-quality samples, specialized equipment, high-performance storage, specialized expertise; limited scalability due to low throughput | Develop low-cost platforms; integrate with other types of data (e.g., scRNA-seq, IHC); expand community of shared datasets and training resources |
| Low accessibility for clinical use | Lack of reproducible, standardized pipelines | Develop user-friendly and automated software for ST data analysis and translation to clinical settings; automate ST platforms for faster clinical workflows |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Park, H.-J.; Kim, M.S. Spatial Transcriptomics in Thyroid Cancer: Applications, Limitations, and Future Perspectives. Cells 2025, 14, 936. https://doi.org/10.3390/cells14120936
Song C, Park H-J, Kim MS. Spatial Transcriptomics in Thyroid Cancer: Applications, Limitations, and Future Perspectives. Cells. 2025; 14(12):936. https://doi.org/10.3390/cells14120936
Chicago/Turabian StyleSong, Chaerim, Hye-Ji Park, and Man S. Kim. 2025. "Spatial Transcriptomics in Thyroid Cancer: Applications, Limitations, and Future Perspectives" Cells 14, no. 12: 936. https://doi.org/10.3390/cells14120936
APA StyleSong, C., Park, H.-J., & Kim, M. S. (2025). Spatial Transcriptomics in Thyroid Cancer: Applications, Limitations, and Future Perspectives. Cells, 14(12), 936. https://doi.org/10.3390/cells14120936