

Free Methylglyoxal and Lactate Produced and Released by Cultured Cancer and Non-Cancer Cells: Implications for Tumor Growth and Development

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Cultures
2.2. Use of Anti-MG Specific Probe
2.3. Free MG and Lactate Measurements
2.4. Statistical Tests
3. Results
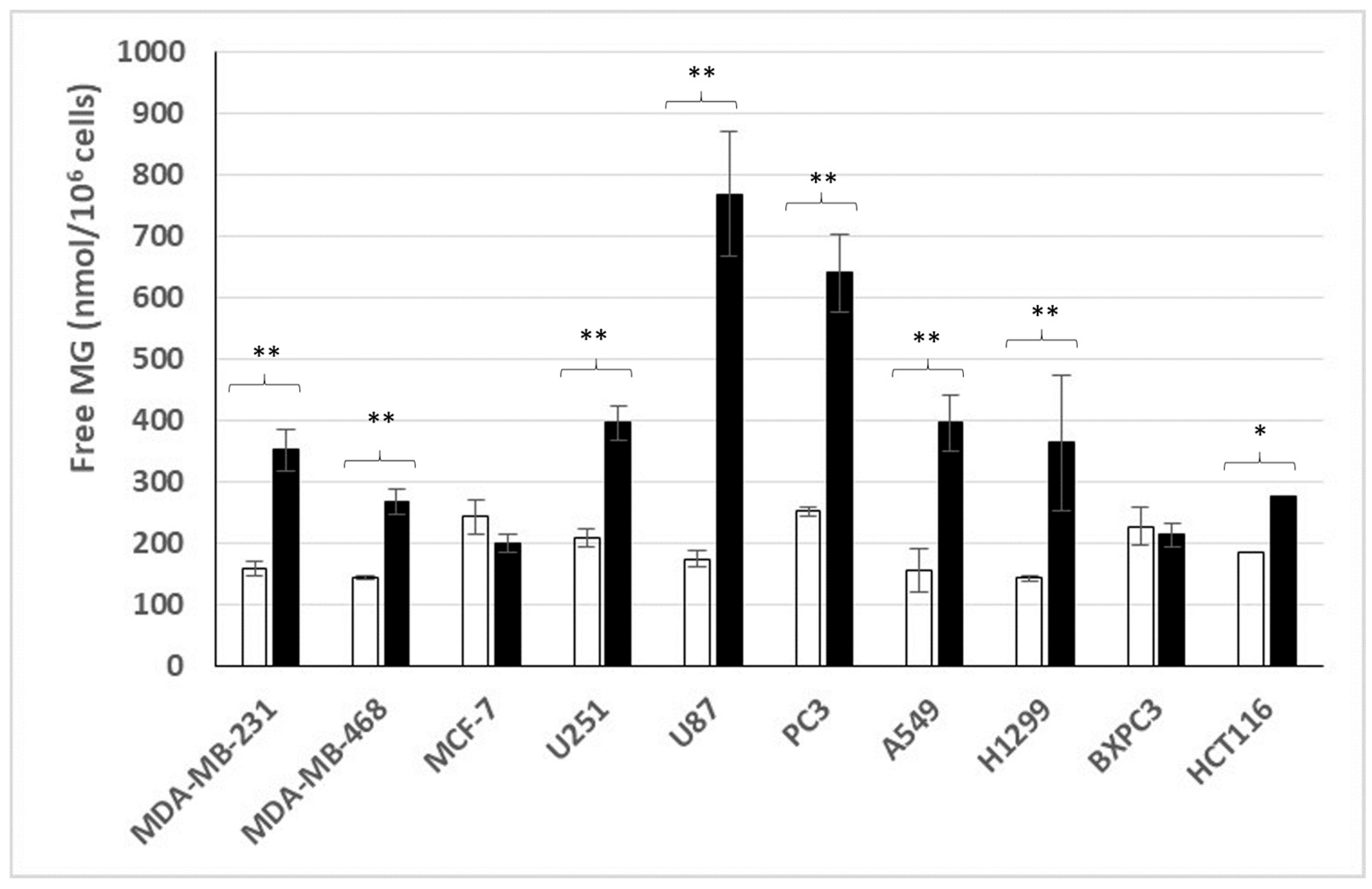
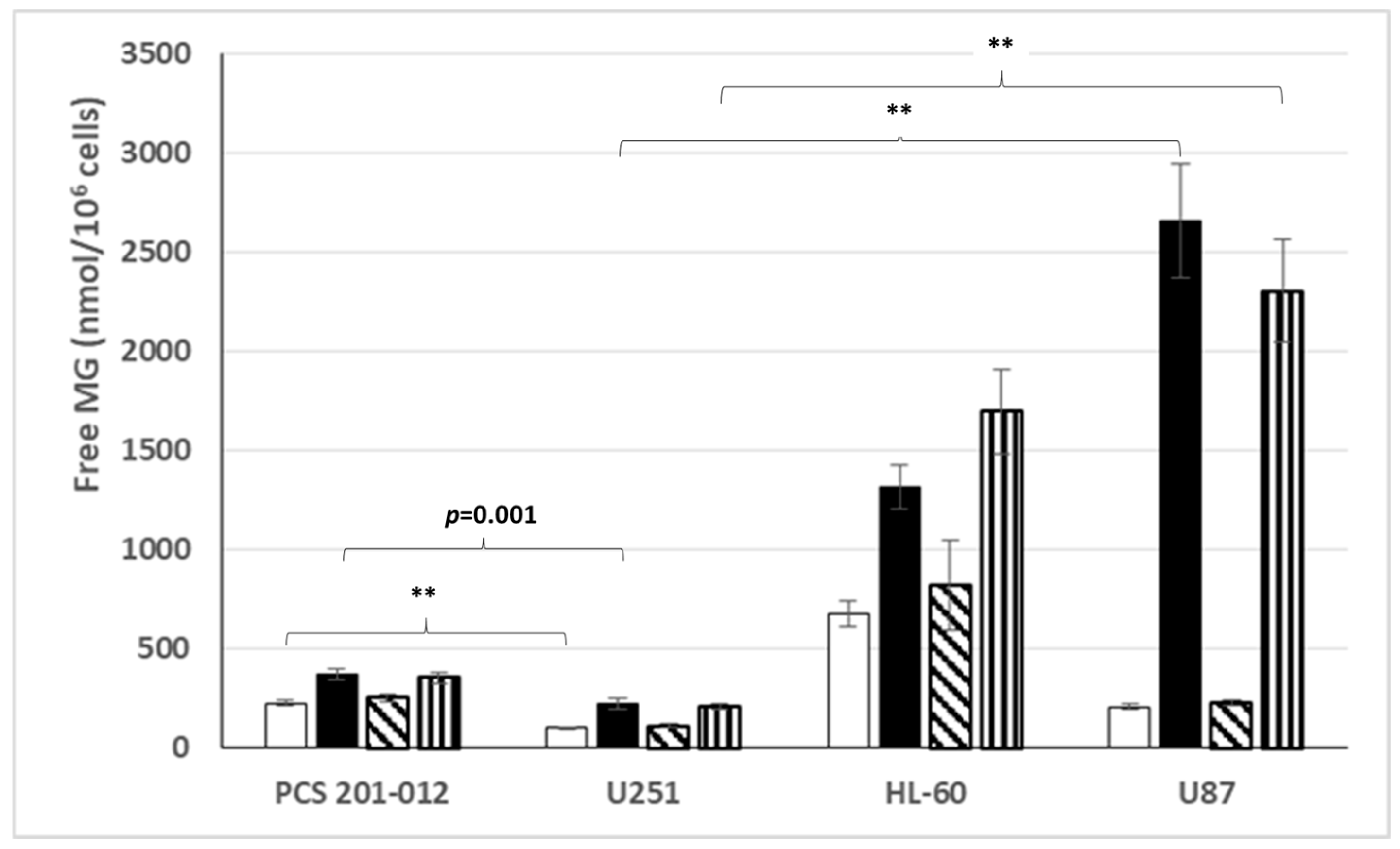
3.1. Free MG Production and Release by Cancer Cell Lines Cultured in Normoxic Low or High Glucose Concentration
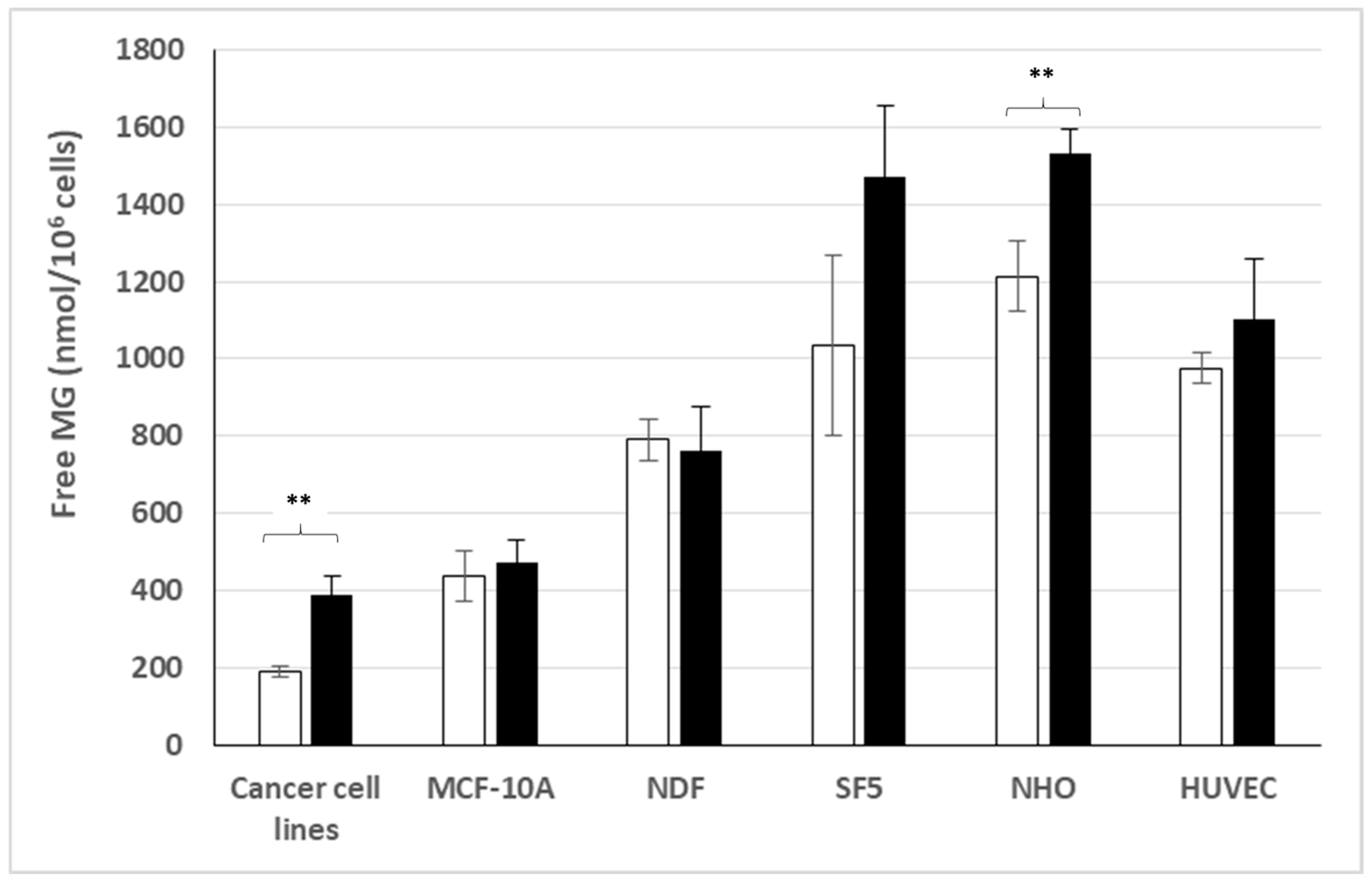
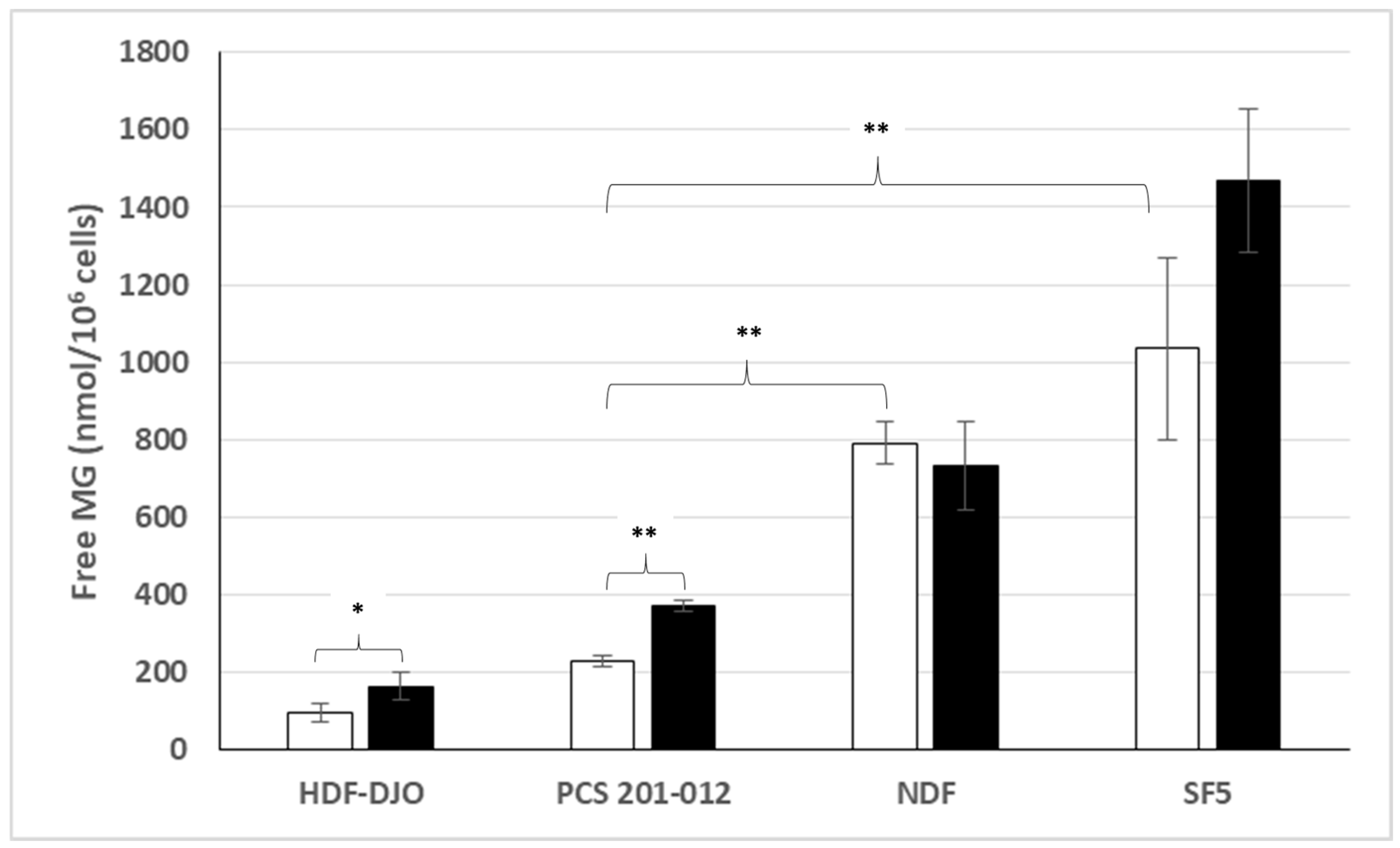
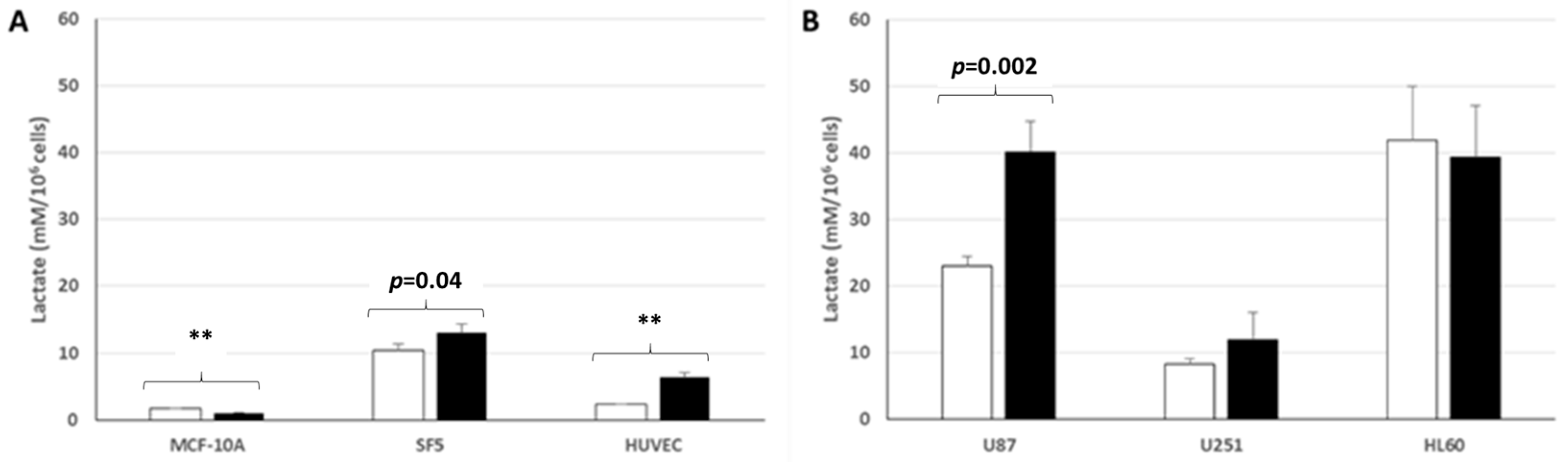
3.2. Free MG Production and Release by Normal Cell Lines Cultured in Normoxic Low or High Glucose Concentration
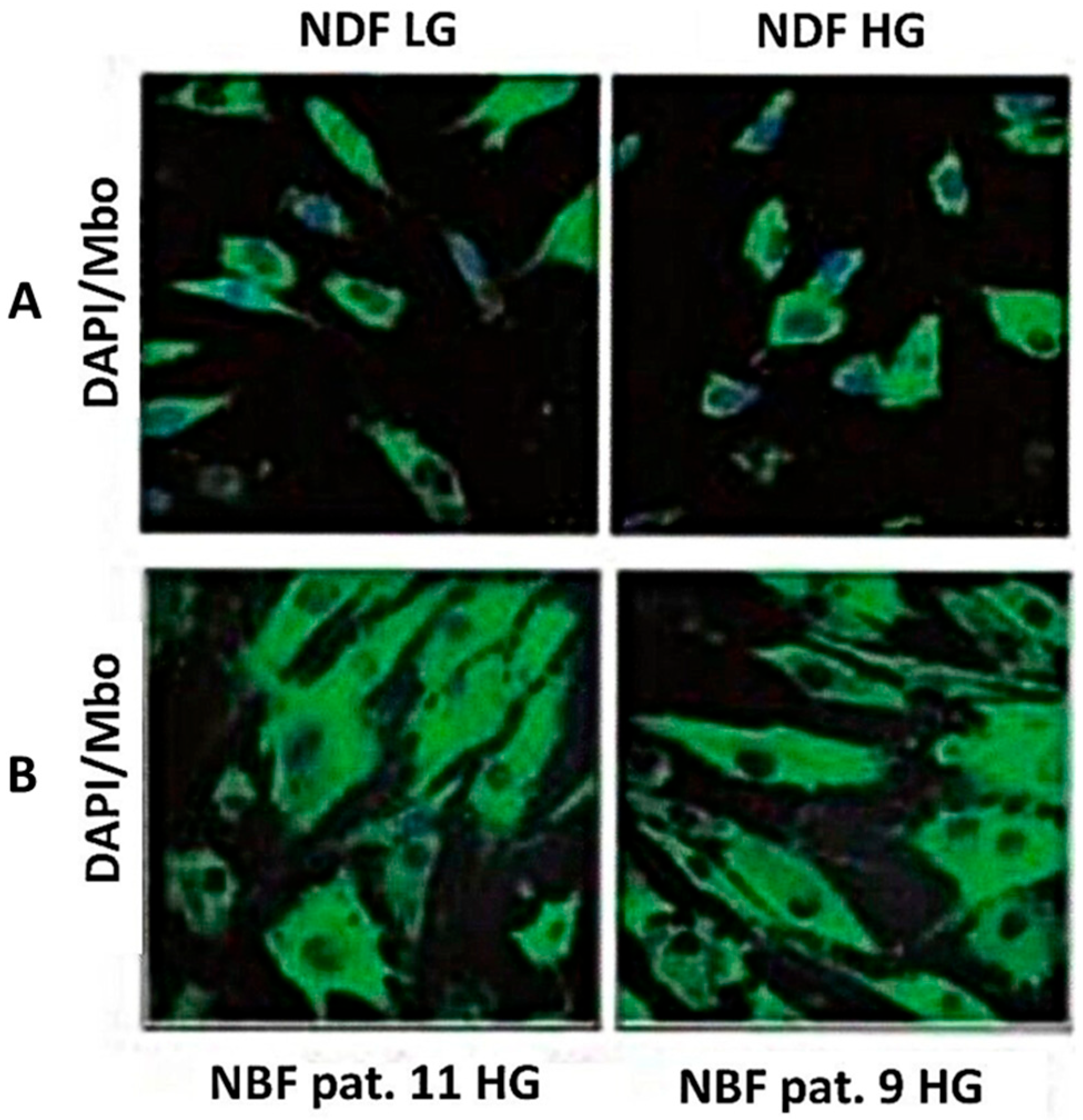
3.3. Intracellular MG Production and Free MG and Lactate Release by Non-Transformed Normal Fibroblasts
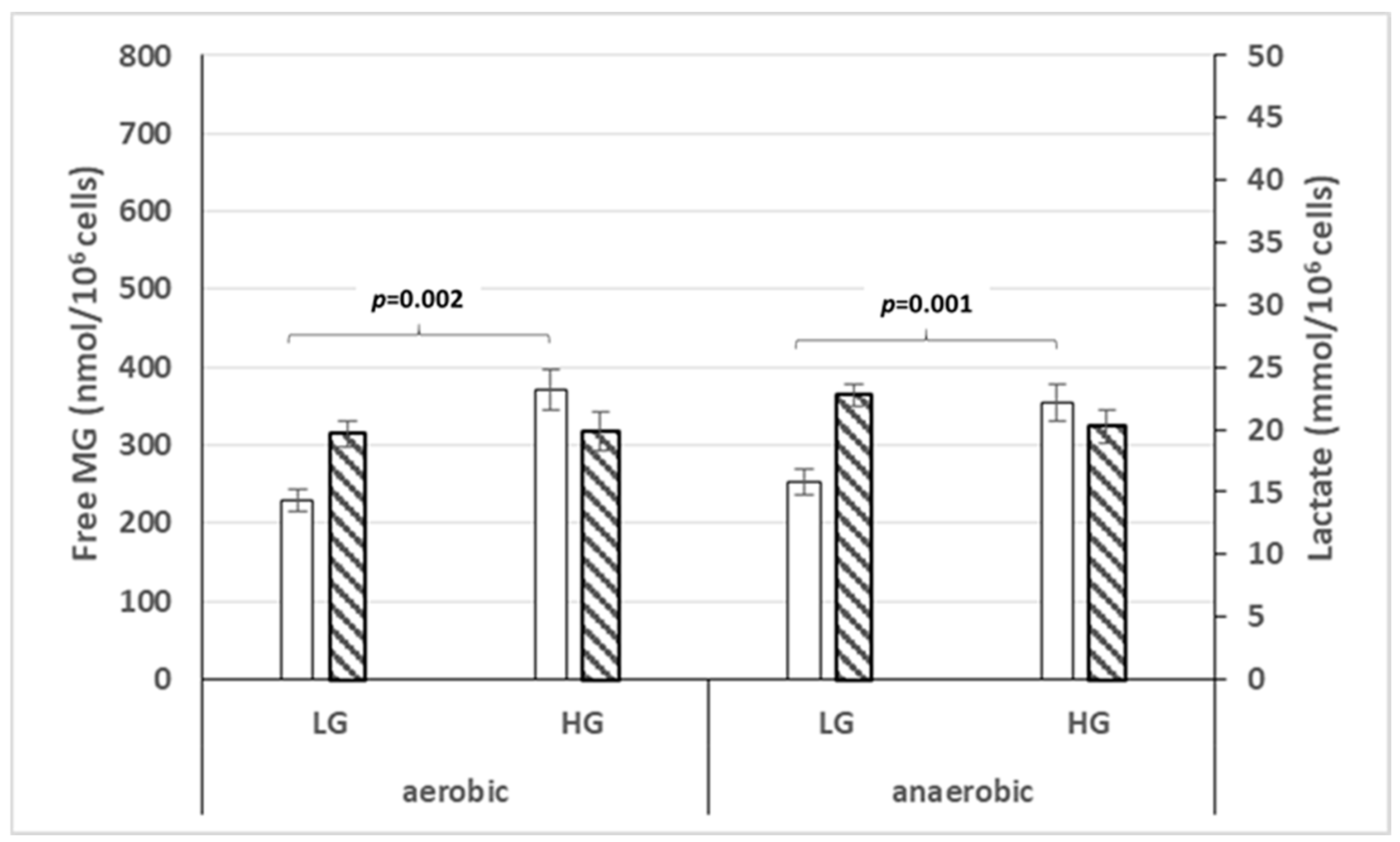
3.4. Free MG Production and Release by Normal Fibroblasts and Cancer Cell Lines Cultured in Normoxic or Hypoxic Conditions in Low or High Glucose Concentration
3.5. Lactate Production and Release by Normal and Cancer Cell Lines Cultured in Normoxic Low or High Glucose Concentration
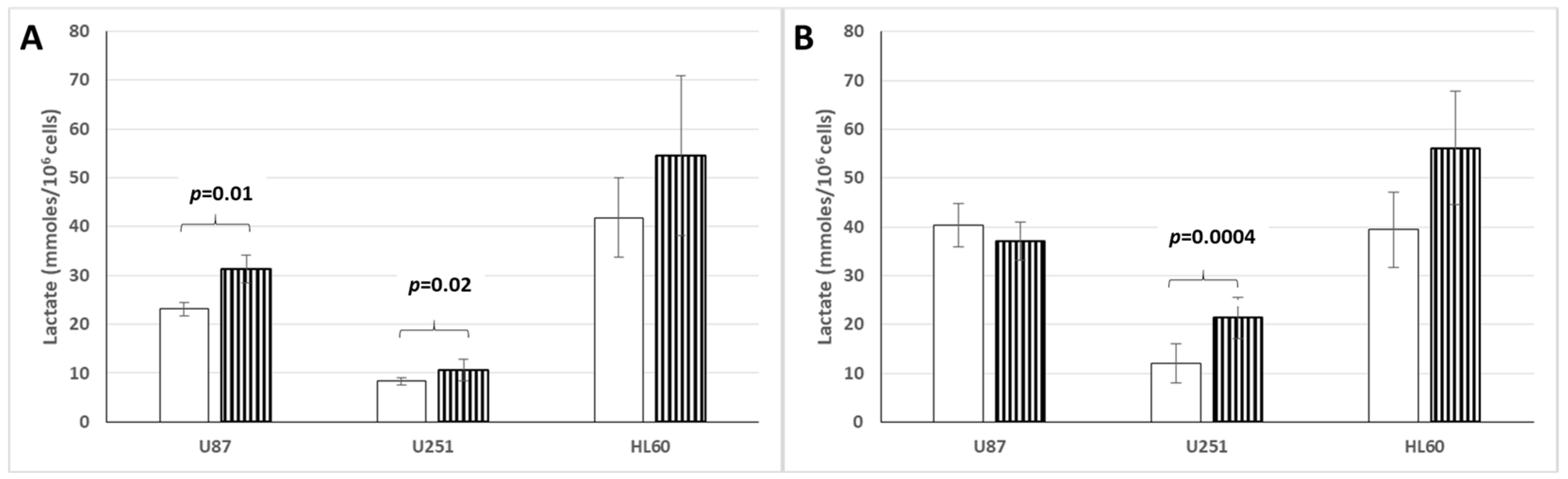
3.6. Lactate Production and Release by Cancer Cell Lines Cultured in Hypoxic or Normoxic Conditions and Low or High Glucose Concentration
4. Discussion
4.1. Free MG Production by Cancer Cells and Non-Cancer Cells
4.2. Free MG and Lactate as Permeable and Diffusible Molecules
4.3. On the Non-Specificity of the Warburg Effect Applied to Isolated Cultured Cells
4.4. High Methylglyoxal Production as a New Core Hallmark of Cell Metabolic Reprogramming in Cancer
4.5. Free MG and Lactate as Intra-Tumoral Molecular Effectors That May Contribute Distinctively to Tumor Growth and Development
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vascular Reactions of Normal and Malignant Tissues in Vivo. I. Vascular Reactions of Mice to Wounds and to Normal and Neoplastic Transplants Get access Arrow. JNCI 1945, 3, 73–85. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis. Adv. Cancer Res. 1974, 19, 331–358. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Lemons, J.M.; Feng, X.J.; Bennett, B.D.; Legesse-Miller, A.; Johnson, E.L.; Raitman, I.; Pollina, E.A.; Rabitz, H.A.; Rabinowitz, J.D.; Coller, H.A. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010, 8, e1000514. [Google Scholar] [CrossRef]
- Durand, R.E.; Raleigh, J.A. Identification of nonproliferating but viable hypoxic tumor cells in vivo. Cancer Res. 1998, 58, 3547–3550. [Google Scholar]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- West, C.M.; Cooper, R.A.; Loncaster, J.A.; Wilks, D.P.; Bromley, M. Tumor vascularity: A histological measure of angiogenesis and hypoxia. Cancer Res. 2001, 61, 2907–2910. [Google Scholar] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Kroemer, G. Essential role for oxidative phosphorylation in cancer progression. Cell Metab. 2015, 21, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef]
- Paul, S.; Ghosh, S.; Kumar, S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin. Cancer Biol. 2022, 86 Pt 3, 1216–1230. [Google Scholar] [CrossRef]
- Ohmori, S.; Mori, M.; Shiraha, K.; Kawase, M. Biosynthesis and degradation of methylglyoxal in animals. Prog. Clin. Biol. Res. 1989, 290, 397–412. [Google Scholar]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. The dicarbonyl proteome: Proteins susceptible to dicarbonyl glycation at functional sites in health, aging, and disease. Ann. N. Y. Acad. Sci. 2008, 1126, 124–127. [Google Scholar] [CrossRef]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification—A role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993, 14, 287–371. [Google Scholar] [CrossRef] [PubMed]
- Caignard, A.; Pelletier, H.; Martin, F. Specificity of the immune response leading to protection or enhancement by regressive and progressive variants of a rat colon carcinoma. Int. J. Cancer 1988, 42, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Irigaray, P.; Belpomme, D. Circulating free methylglyoxal as a metabolic tumor biomarker in a rat colon adenocarcinoma model. Mol. Clin. Oncol. 2020, 12, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Belpomme, D.; Lacomme, S.; Poletti, C.; Bonesso, L.; Hinault-Boyer, C.; Barbier, S.; Irigaray, P. Free Methylglyoxal as a Metabolic New Biomarker of Tumor Cell Proliferation in Cancers. Cancers 2024, 16, 3922. [Google Scholar] [CrossRef]
- Chaplen, F.W.; Fahl, W.E.; Cameron, D.C. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal. Biochem. 1996, 238, 171–178. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Kong, X.; Ma, M.Z.; Huang, K.; Qin, L.; Zhang, H.M.; Yang, Z.; Li, X.Y.; Su, Q. Increased plasma levels of the methylglyoxal in patients with newly diagnosed type 2 diabetes 2. J. Diabetes 2014, 6, 535–540. [Google Scholar] [CrossRef]
- Wang, T.; Douglass, E.F., Jr.; Fitzgerald, K.J.; Spiegel, D.A. A “turn-on” fluorescent sensor for methylglyoxal. J. Am. Chem. Soc. 2013, 135, 12429–12433. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 2014, 9, 1969–1979. [Google Scholar] [CrossRef]
- Tan, B.; Lu, Z.; Dong, S.; Zhao, G.; Kuo, M.S. Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal. Biochem. 2014, 465, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Lee, H.J.; Howell, S.K.; Sanford, R.J.; Beisswenger, P.J. Methylglyoxal can modify GAPDH activity and structure. Ann. N. Y. Acad. Sci. 2005, 1043, 135–145. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.D.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef]
- Leone, A.; Nigro, C.; Nicolò, A.; Prevenzano, I.; Formisano, P.; Beguinot, F.; Miele, C. The Dual-Role of Methylglyoxal in Tumor Progression—Novel Therapeutic Approaches. Front. Oncol. 2021, 11, 645686. [Google Scholar] [CrossRef] [PubMed]
- Dolce, V.; Cappello, A.R.; Capobianco, L. Mitochondrial tricarboxylate and dicarboxylate-tricarboxylate carriers: From animals to plants. IUBMB Life 2014, 66, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Harmati, M.; Bukva, M.; Böröczky, T.; Buzás, K.; Gyukity-Sebestyén, E. The role of the metabolite cargo of extracellular vesicles in tumor progression. Cancer Metastasis Rev. 2021, 40, 1203–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, F.; Zhang, D.; Qi, S.; Liu, Y. Metabolites as extracellular vesicle cargo in health, cancer, pleural effusion, and cardiovascular diseases: An emerging field of study to diagnostic and therapeutic purposes. Biomed. Pharmacother. 2023, 157, 114046. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Jerzykowski, T.; Winter, R.; Matuszewski, W.; Piskorska, D. A re-evaluation of studies on the distribution of glyoxalases in animal and tumour tissues. Int. J. Biochem. 1978, 9, 853–860. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23 (6 Pt B), 522–532. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Scoltock, A.B.; Cain, D.W.; Cidlowski, J.A. T-cell development of resistance to apoptosis is driven by a metabolic shift in carbon source and altered activation of death pathways. Cell Death Differ. 2016, 23, 889–902. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef]
- Di Ilio, C.; Angelucci, S.; Pennelli, A.; Zezza, A.; Tenaglia, R.; Sacchetta, P. Glyoxalase activities in tumor and non-tumor human urogenital tissues. Cancer Lett. 1995, 96, 189–193. [Google Scholar] [CrossRef]
- Dragani, B.; Cocco, R.; Ridderström, M.; Stenberg, G.; Mannervik, B.; Aceto, A. Unfolding and refolding of human glyoxalase II and its single-tryptophan mutants. J. Mol. Biol. 1999, 291, 481–490. [Google Scholar] [CrossRef]
- Santel, T.; Pflug, G.; Hemdan, N.Y.; Schäfer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef]
- de Bari, L.; Moro, L.; Passarella, S. Prostate cancer cells metabolize d-lactate inside mitochondria via a D-lactate dehydrogenase which is more active and highly expressed than in normal cells. FEBS Lett. 2013, 587, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Zhou, B.W.; Cao, Y.Q.; Zhang, J.; Zhang, L.; Guo, Y.L. Chiral Analysis of Lactate during Direct Contact Coculture by Single-Cell On-Probe Enzymatic Dehydrogenation Derivatization: Unraveling Metabolic Changes Caused by d-Lactate. Anal. Chem. 2021, 93, 4576–4583. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Flick, M.J.; Konieczny, S.F. Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem. Biophys. Res. Commun. 2002, 295, 910–916. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Wan, F.; Dai, B.; Ye, D. Prognostic value of D-lactate dehydrogenase in patients with clear cell renal cell carcinoma. Oncol. Lett. 2018, 16, 866–874. [Google Scholar] [CrossRef]
- Dennis, D.; Kaplan, N.O. D- and L-lactic acid dehydrogenases in Lactobacillus plantarum. J. Biol. Chem. 1960, 235, 810–818. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate shuttles in nature. Biochem. Soc. Trans. 2002, 30, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lyons, J.C.; Ohtsubo, T.; Song, C.W. Acidic environment causes apoptosis by increasing caspase activity. Br. J. Cancer 1999, 80, 1892–1897. [Google Scholar] [CrossRef]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Gillies, R.J.; Gatenby, R.A. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007, 26, 311–317. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Miki, S.; Kasayama, S.; Miki, Y.; Nakamura, Y.; Yamamoto, M.; Sato, B.; Kishimoto, T. Expression of receptors for advanced glycosylation end products on renal cell carcinoma cells in vitro. Biochem. Biophys. Res. Commun. 1993, 196, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced glycation end product recognition by the receptor for AGEs. Structure 2011, 19, 722–732. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, K.C.; Chang, G.C.; Lin, H.; Wu, C.C.; Kao, W.H.; Teng, C.J.; Hsu, S.L.; Yang, T.Y. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Hammond, N.G.; Cameron, R.B.; Faubert, B. Beyond glucose and Warburg: Finding the sweet spot in cancer metabolism models. NPJ Metab Health Dis. 2024, 2, 11. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belpomme, D.; Irigaray, P.; Alberto, J.-M.; Poletti, C.; Hinault-Boyer, C.; Lacomme, S. Free Methylglyoxal and Lactate Produced and Released by Cultured Cancer and Non-Cancer Cells: Implications for Tumor Growth and Development. Cells 2025, 14, 931. https://doi.org/10.3390/cells14120931
Belpomme D, Irigaray P, Alberto J-M, Poletti C, Hinault-Boyer C, Lacomme S. Free Methylglyoxal and Lactate Produced and Released by Cultured Cancer and Non-Cancer Cells: Implications for Tumor Growth and Development. Cells. 2025; 14(12):931. https://doi.org/10.3390/cells14120931
Chicago/Turabian StyleBelpomme, Dominique, Philippe Irigaray, Jean-Marc Alberto, Clément Poletti, Charlotte Hinault-Boyer, and Stéphanie Lacomme. 2025. "Free Methylglyoxal and Lactate Produced and Released by Cultured Cancer and Non-Cancer Cells: Implications for Tumor Growth and Development" Cells 14, no. 12: 931. https://doi.org/10.3390/cells14120931
APA StyleBelpomme, D., Irigaray, P., Alberto, J.-M., Poletti, C., Hinault-Boyer, C., & Lacomme, S. (2025). Free Methylglyoxal and Lactate Produced and Released by Cultured Cancer and Non-Cancer Cells: Implications for Tumor Growth and Development. Cells, 14(12), 931. https://doi.org/10.3390/cells14120931