
Inter-Relationship Between Melanoma Vemurafenib Tolerance Thresholds and Metabolic Pathway Choice

 , ,
, ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cell Survival and Colony-Forming Analyses
2.3. Western Blot Analysis
2.4. Chemotaxis Analysis
2.5. Dual-Luciferase Reporter Assay
2.6. Mitochondria Imaging Analysis
2.7. Analysis of Sugar Utilization Flexibility
2.8. Cellular Bioenergetics Analysis
2.9. Immunofluorescence Staining
2.10. Targeted Metabolomics
2.11. Mitochondrial and Cytosol Metabolism Analysis
2.12. Whole Genome Expression Analysis by RNA-Seq
2.13. Real-Time Reverse Transcriptase (RT)-PCR Analysis
2.14. Statistical Analysis
3. Results
3.1. Generation and Characterization of Isogenic Models of Vemurafenib Resistant (VemR) Melanoma Cells
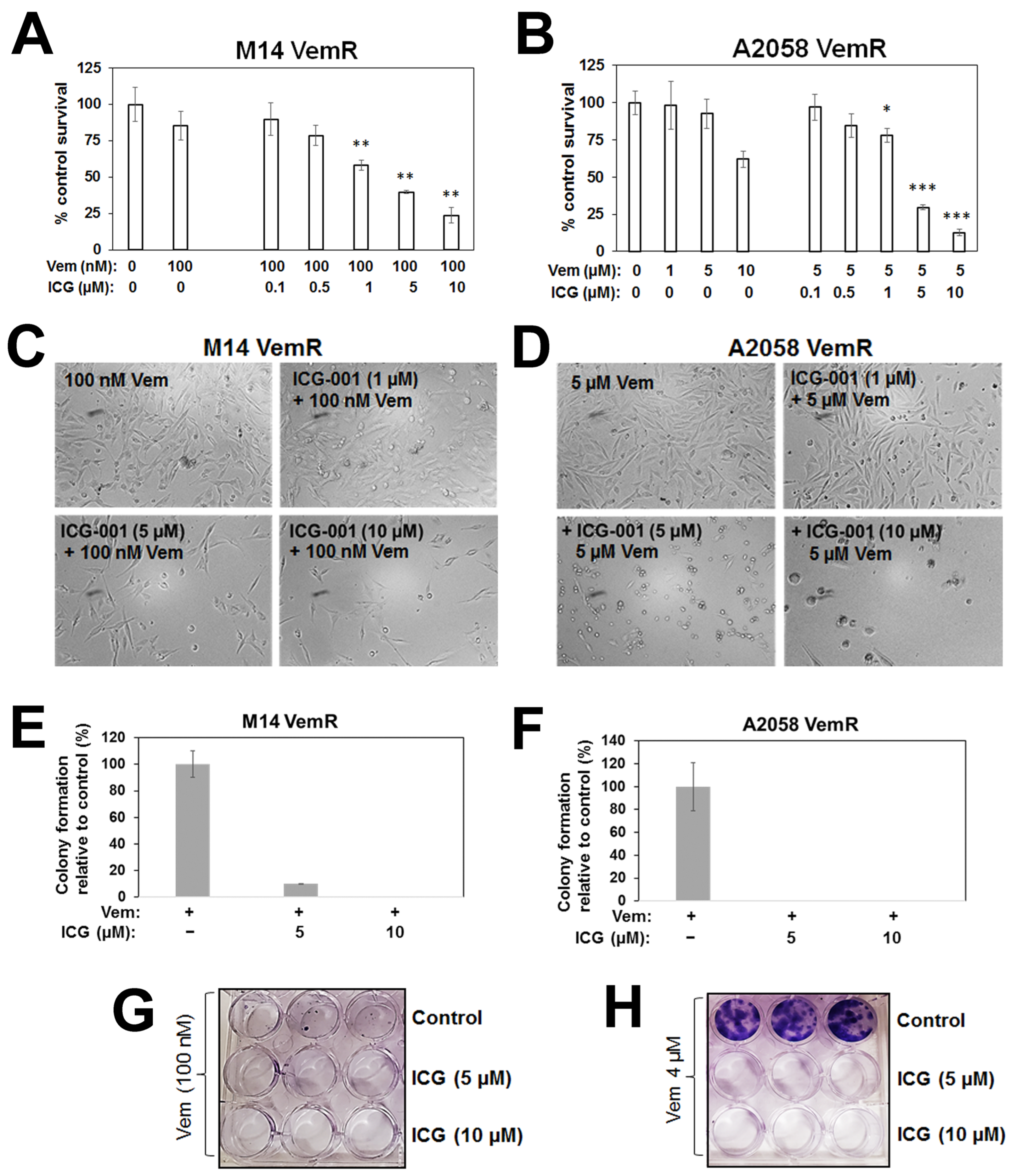
3.2. β-Catenin Transcriptional Activity Inhibition Sensitizes Vemurafenib Resistant Melanoma Cells
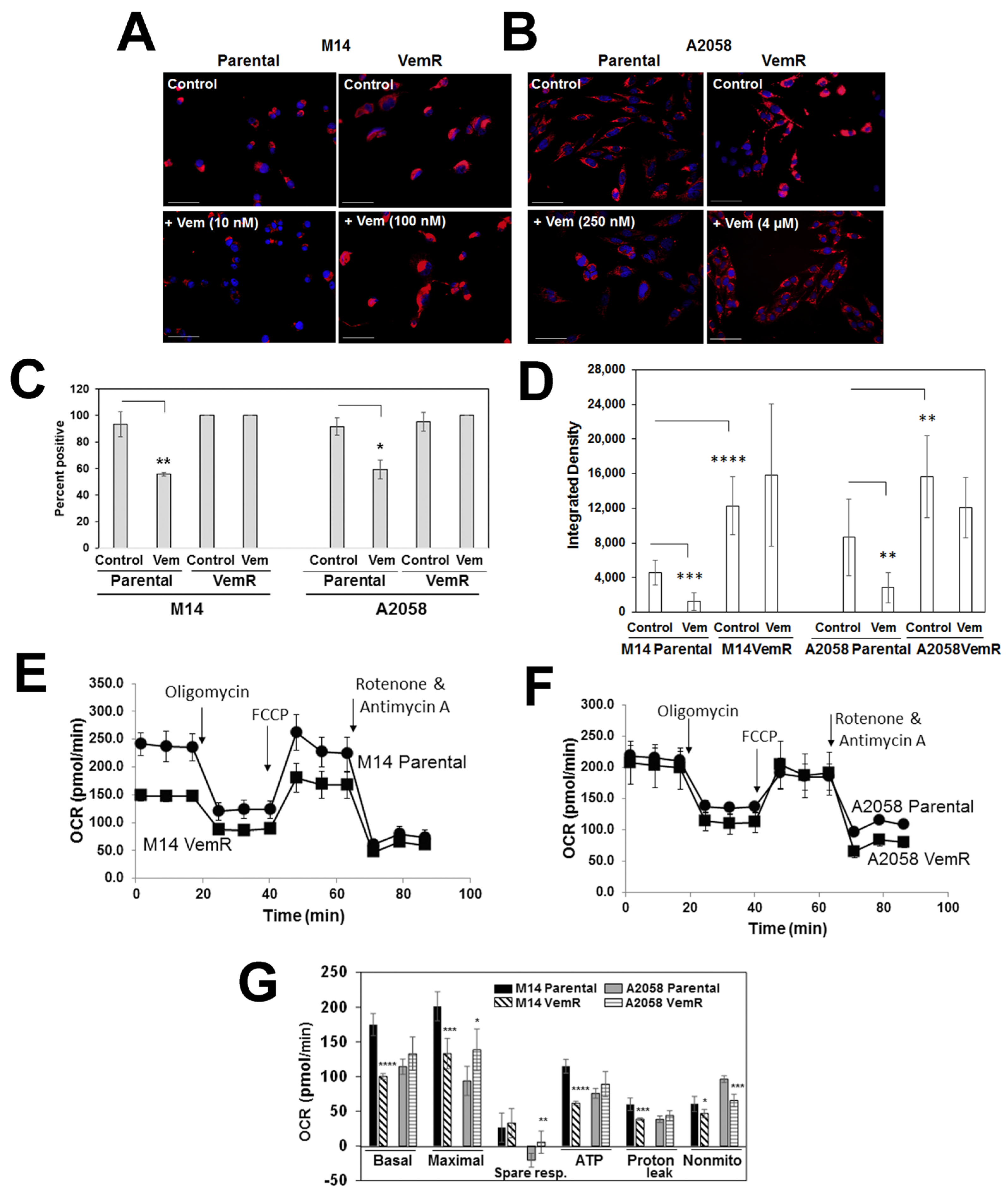
3.3. VemR Melanoma Cells Have Elevated Levels of Mitochondrial Mass and OXPHOS Activity
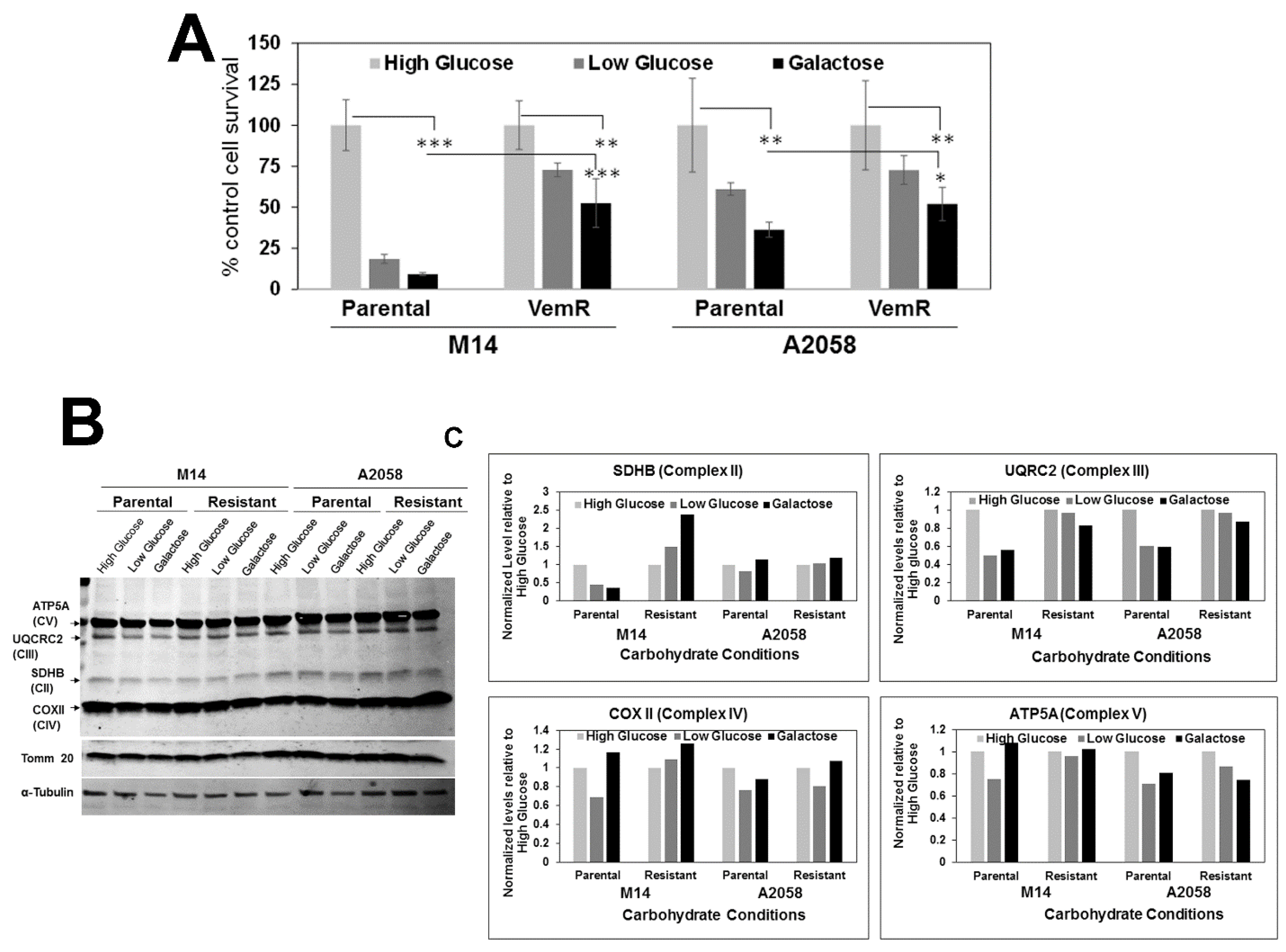
3.4. VemR Melanoma Cells Have Increased Abilities to Utilize Galactose as the Sole Energy Source
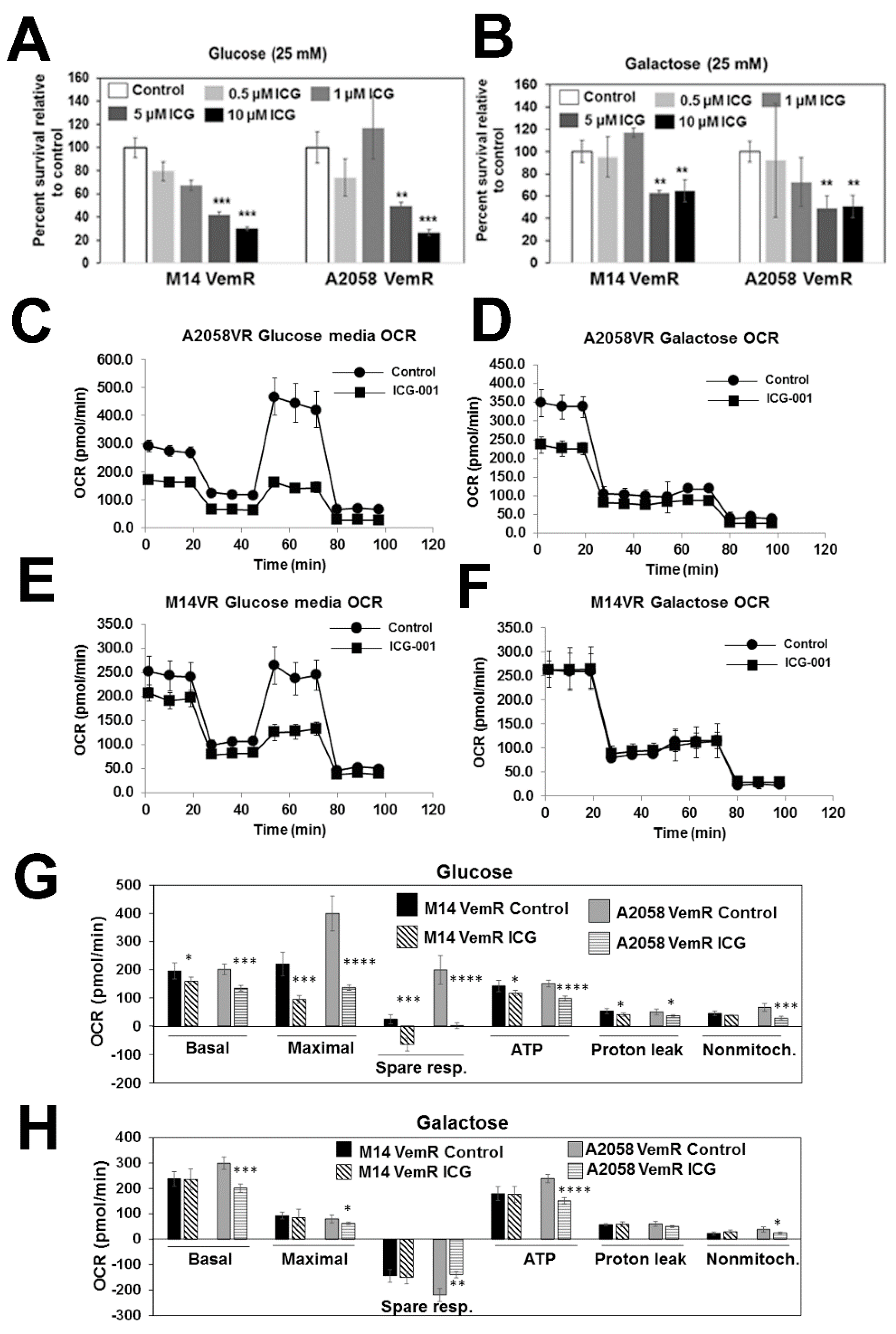
3.5. β-Catenin Signaling Promotes Mitochondrial Bioenergetics in VemR Melanoma Cells
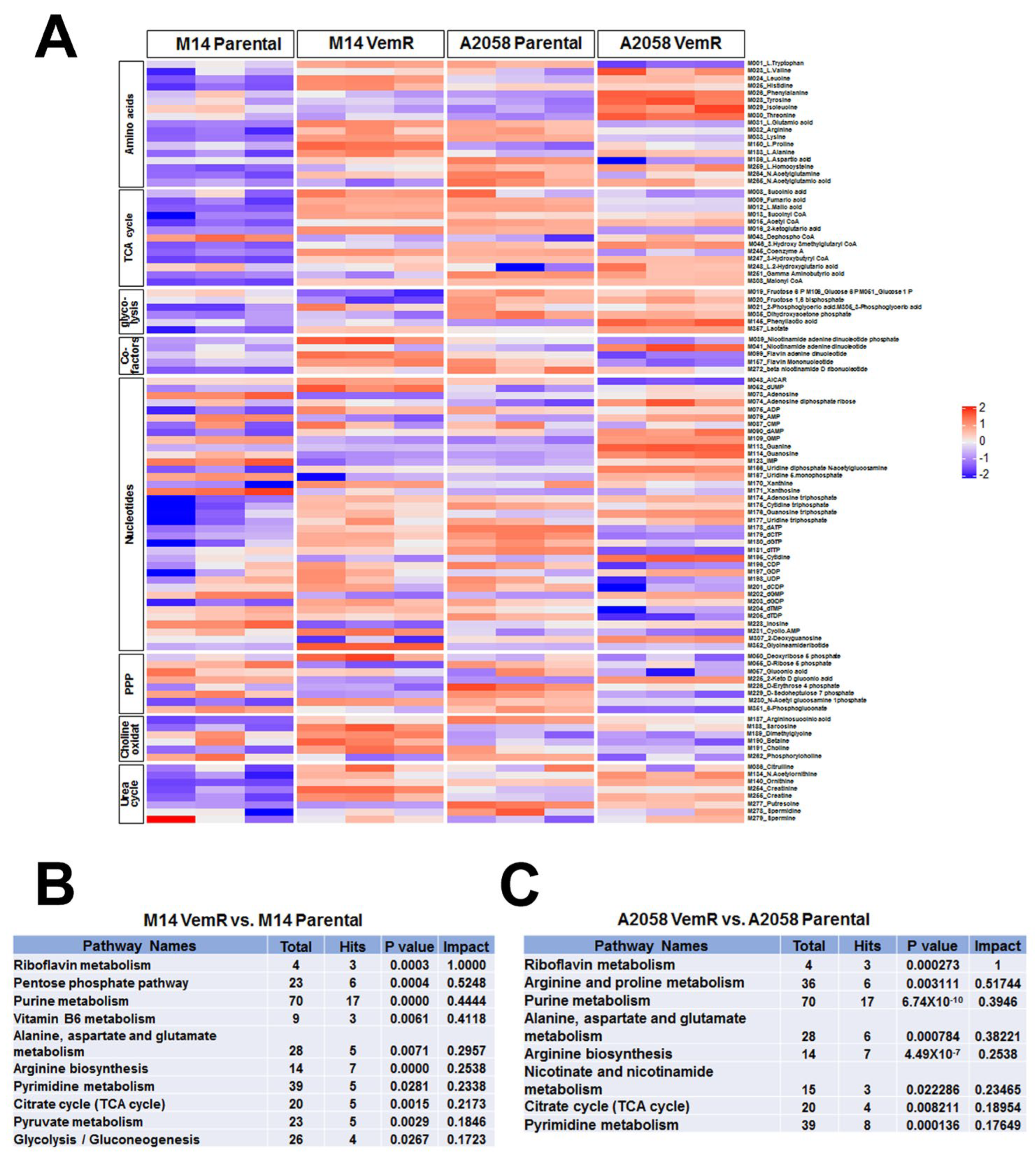
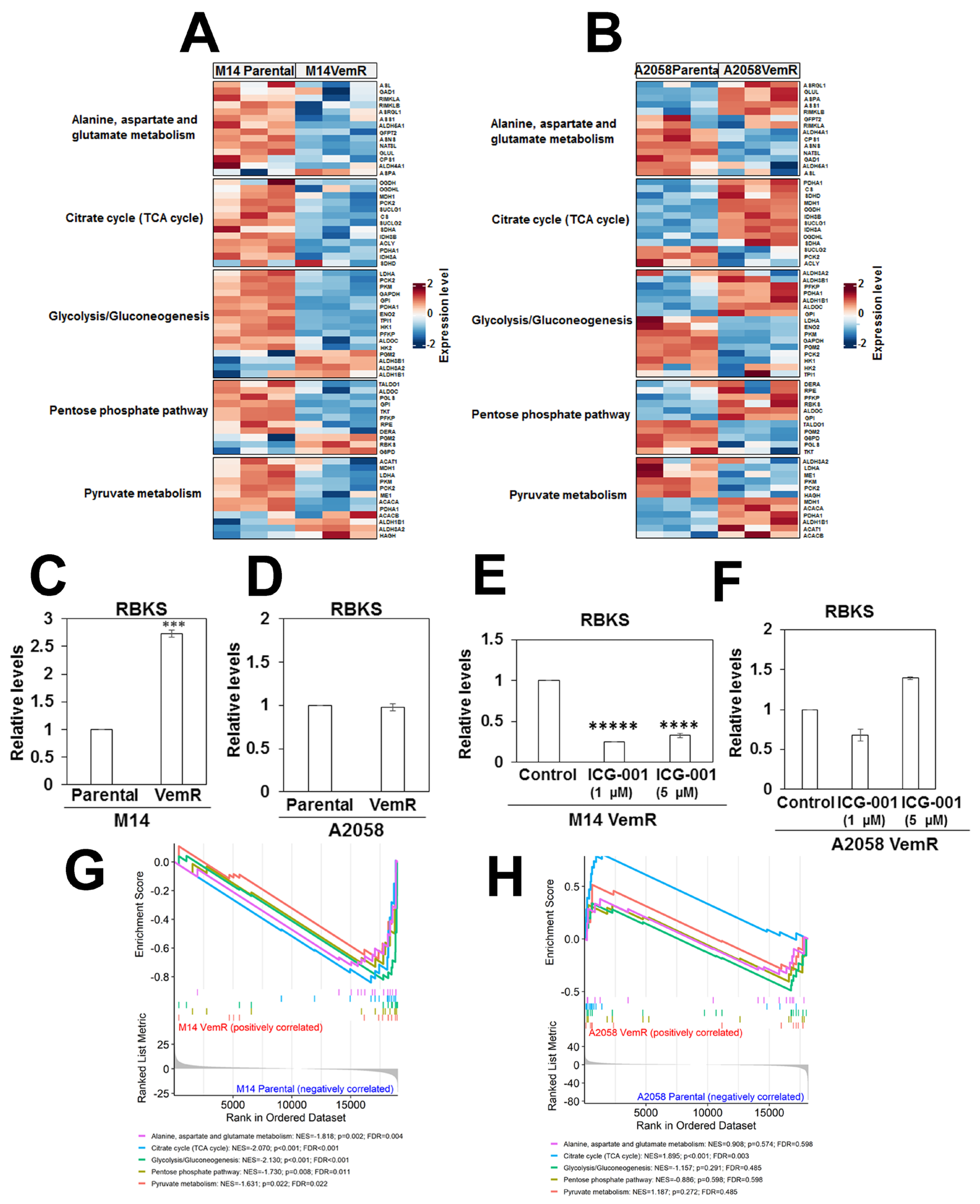
3.6. Vemurafenib Resistance Development Is Associated with Alterations in Metabolic Pathways Involved with TCA Cycle and Pentose Phosphate Pathway
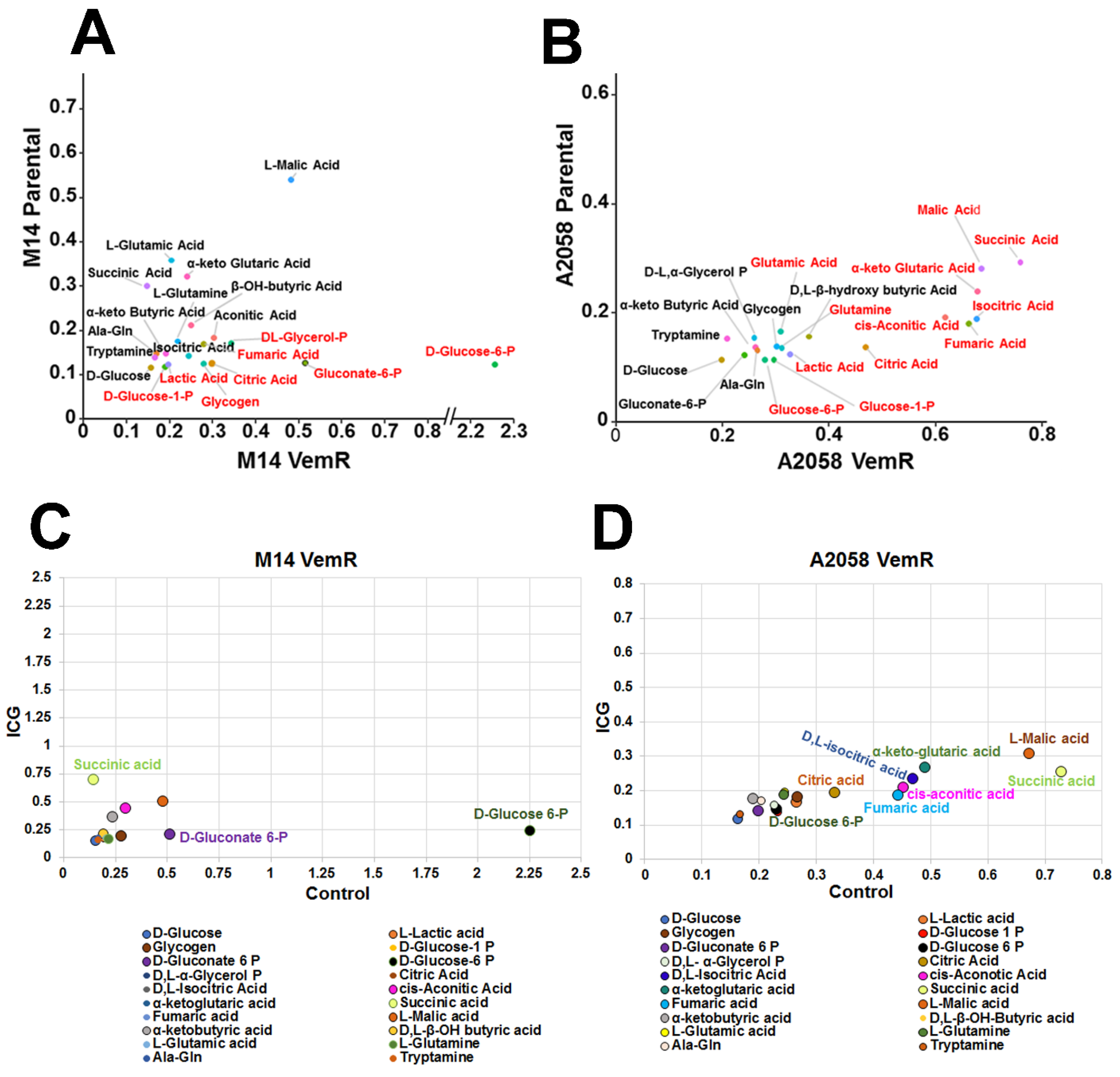
3.7. VemR Melanoma Cells Have Increased Ability to Metabolize Mitochondrial Substrates to Generate ATP
3.8. Transcriptome Analysis Supports VemR-Associated Alterations in β-Catenin Signaling and Metabolic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caraban, B.M.; Aschie, M.; Deacu, M.; Cozaru, G.C.; Pundiche, M.B.; Orasanu, C.I.; Voda, R.I. A Narrative Review of Current Knowledge on Cutaneous Melanoma. Clin. Pract. 2024, 14, 214–241. [Google Scholar] [CrossRef] [PubMed]
- Giunta, E.F.; De Falco, V.; Napolitano, S.; Argenziano, G.; Brancaccio, G.; Moscarella, E.; Ciardiello, D.; Ciardiello, F.; Troiani, T. Optimal treatment strategy for metastatic melanoma patients harboring BRAF-V600 mutations. Ther. Adv. Med. Oncol. 2020, 12, 1758835920925219. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.E.; Torrisi, E.; Bevelacqua, Y.; Perrotta, R.; Libra, M.; McCubrey, J.A.; Spandidos, D.A.; Stivala, F.; Malaponte, G. Melanoma: Molecular pathogenesis and emerging target therapies. Int. J. Oncol. 2009, 34, 1481–1489. [Google Scholar] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E) BRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E.; Widlund, H.R. Molecular pathways. BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin. Cancer Res. 2014, 20, 2257–2263. [Google Scholar]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Kim, Y.M.; Kahn, M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. Rep. 2014, 10, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Spranger, S.; Fuchs, E.; Lopez-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65. [Google Scholar] [CrossRef]
- Vance, K.W.; Goding, C.R. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res. 2004, 17, 318–325. [Google Scholar] [CrossRef]
- Ngeow, K.C.; Friedrichsen, H.J.; Li, L.; Zeng, Z.; Andrews, S.; Volpon, L.; Brunsdon, H.; Berridge, G.; Picaud, S.; Fischer, R.; et al. BRAF/MAPK and GSK3 signaling converges to control MITF nuclear export. Proc. Natl. Acad. Sci. USA 2018, 115, E8668–E8677. [Google Scholar] [CrossRef]
- Sarma, A.; Gajan, A.; Kim, S.; Gurdziel, K.; Mao, G.; Nangia-Makker, P.; Shekhar, M.P.V. RAD6B Loss Disrupts Expression of Melanoma Phenotype in Part by Inhibiting WNT/beta-Catenin Signaling. Am. J. Pathol. 2021, 191, 368–384. [Google Scholar] [CrossRef]
- Gajan, A.; Martin, C.E.; Kim, S.; Joshi, M.; Michelhaugh, S.K.; Sloma, I.; Mittal, S.; Firestine, S.; Shekhar, M.P.V. Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma. Identification and Functional Characterization. Cells 2019, 8, 1375. [Google Scholar] [CrossRef]
- Shekhar, M.P.; Tait, L.; Gerard, B. Essential role of T-cell factor/beta-catenin in regulation of Rad6B. A potential mechanism for Rad6B overexpression in breast cancer cells. Mol. Cancer Res. 2006, 4, 729–745. [Google Scholar] [CrossRef]
- Yue, Y.; Bao, X.; Jiang, J.; Li, J. Evaluation and correction of injection order effects in LC-MS/MS based targeted metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2022, 1212, 123513. [Google Scholar] [CrossRef]
- Pang, Z.; Xu, L.; Viau, C.; Lu, Y.; Salavati, R.; Basu, N.; Xia, J. MetaboAnalystR 4.0. a unified LC-MS workflow for global metabolomics. Nat. Commun. 2024, 15, 3675. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR. ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR. a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate. A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Fratta, E.; Giurato, G.; Guerrieri, R.; Colizzi, F.; Dal Col, J.; Weisz, A.; Steffan, A.; Montico, B. Autophagy in BRAF-mutant cutaneous melanoma. recent advances and therapeutic perspective. Cell Death Discov. 2023, 9, 202. [Google Scholar] [CrossRef]
- Chen, G.; Gao, C.; Gao, X.; Zhang, D.H.; Kuan, S.F.; Burns, T.F.; Hu, J. Wnt/beta-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 806–813. [Google Scholar] [CrossRef]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma—A mini review. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 2011, 6, e28536. [Google Scholar] [CrossRef]
- Dott, W.; Mistry, P.; Wright, J.; Cain, K.; Herbert, K.E. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol. 2014, 2, 224–233. [Google Scholar] [CrossRef]
- Radogna, F.; Gerard, D.; Dicato, M.; Diederich, M. Assessment of Mitochondrial Cell Metabolism by Respiratory Chain Electron Flow Assays. Methods Mol. Biol. 2021, 2276, 129–141. [Google Scholar] [PubMed]
- Kordus, R.J.; Hossain, A.; Malter, H.E.; LaVoie, H.A. Mitochondrial metabolic substrate utilization in granulosa cells reflects body mass index and total follicle stimulating hormone dosage in in vitro fertilization patients. J. Assist. Reprod. Genet. 2020, 37, 2743–2756. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liang, S.; Wasylishen, A.R.; Zhang, Y.; Yang, X.; Zhou, B.; Shan, L.; Han, X.; Mu, T.; Wang, G.; et al. PLA2G16 promotes osteosarcoma metastasis and drug resistance via the MAPK pathway. Oncotarget 2016, 7, 18021–18035. [Google Scholar] [CrossRef] [PubMed]
- Hankey, W.; Chen, Z.; Bergman, M.J.; Fernandez, M.O.; Hancioglu, B.; Lan, X.; Jegga, A.G.; Zhang, J.; Jin, V.X.; Aronow, B.J.; et al. Chromatin-associated APC regulates gene expression in collaboration with canonical WNT signaling and AP-1. Oncotarget 2018, 9, 31214–31230. [Google Scholar] [CrossRef]
- Shi, X.; Cen, Y.; Shan, L.; Tian, L.; Zhu, E.; Yuan, H.; Li, X.; Liu, Y.; Wang, B. N-myc downstream regulated gene 1 suppresses osteoblast differentiation through inactivating Wnt/beta-catenin signaling. Stem Cell Res. Ther. 2022, 13, 53. [Google Scholar] [CrossRef]
- Xiao, J.; Egger, M.E.; McMasters, K.M.; Hao, H. Differential expression of ABCB5 in BRAF inhibitor-resistant melanoma cell lines. BMC Cancer 2018, 18, 675. [Google Scholar] [CrossRef]
- Wilson, B.J.; Saab, K.R.; Ma, J.; Schatton, T.; Putz, P.; Zhan, Q.; Murphy, G.F.; Gasser, M.; Waaga-Gasser, A.M.; Frank, N.Y.; et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014, 74, 4196–4207. [Google Scholar] [CrossRef]
- Louphrasitthiphol, P.; Chauhan, J.; Goding, C.R. ABCB5 is activated by MITF and beta-catenin and is associated with melanoma differentiation. Pigment Cell Melanoma Res. 2020, 33, 112–118. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Corazao-Rozas, P.; Guerreschi, P.; Andre, F.; Gabert, P.E.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef]
- Sherwood, V. WNT signaling: An emerging mediator of cancer cell metabolism? Mol. Cell. Biol. 2015, 35, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Chaurasiya, S.K.; Ekstrom, E.J.; Guilmain, W.; Liu, Q.; Koeck, T.; Brown, K.; Hansson, K.; Agnarsdottir, M.; Bergqvist, M.; et al. WNT5A-mediated beta-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis 2014, 35, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef]
- Brown, K.; Yang, P.; Salvador, D.; Kulikauskas, R.; Ruohola-Baker, H.; Robitaille, A.M.; Chien, A.J.; Moon, R.T.; Sherwood, V. WNT/beta-catenin signaling regulates mitochondrial activity to alter the oncogenic potential of melanoma in a PTEN-dependent manner. Oncogene 2017, 36, 3119–3136. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree effect. replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007, 97, 539–547. [Google Scholar] [CrossRef]
- Larribere, L.; Novak, D.; Wu, H.; Huser, L.; Granados, K.; Orouji, E.; Utikal, J. New role of ID3 in melanoma adaptive drug-resistance. Oncotarget 2017, 8, 110166–110175. [Google Scholar]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genom. Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 promotes melanoma metastasis through enhancing NF-kappaB p65 protein stability. Biochem. Biophys. Res. Commun. 2017, 492, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lutz, N.W.; Banerjee, P.; Wilson, B.J.; Ma, J.; Cozzone, P.J.; Frank, M.H. Expression of Cell-Surface Marker ABCB5 Causes Characteristic Modifications of Glucose, Amino Acid and Phospholipid Metabolism in the G3361 Melanoma-Initiating Cell Line. PLoS ONE 2016, 11, e0161803. [Google Scholar] [CrossRef]
- Gerard, L.; Duvivier, L.; Fourrez, M.; Salazar, P.; Sprimont, L.; Xia, D.; Ambudkar, S.V.; Gottesman, M.M.; Gillet, J.P. Identification of two novel heterodimeric ABC transporters in melanoma: ABCB5beta/B6 and ABCB5beta/B9. J. Biol. Chem. 2024, 300, 105594. [Google Scholar]
- Wei, L.; Lee, D.; Law, C.T.; Zhang, M.S.; Shen, J.; Chin, D.W.; Zhang, A.; Tsang, F.H.; Wong, C.L.; Ng, I.O.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef]
- Ramos-Montoya, A.; Lee, W.N.; Bassilian, S.; Lim, S.; Trebukhina, R.V.; Kazhyna, M.V.; Ciudad, C.J.; Noe, V.; Centelles, J.J.; Cascante, M. Pentose phosphate cycle oxidative and nonoxidative balance. A new vulnerable target for overcoming drug resistance in cancer. Int. J. Cancer 2006, 119, 2733–2741. [Google Scholar] [CrossRef]
- Giannecchini, M.; Matteucci, M.; Pesi, R.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Uptake and utilization of nucleosides for energy repletion. Int. J. Biochem. Cell Biol. 2005, 37, 797–808. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Camici, M.; Mascia, L.; Sgarrella, F.; Ipata, P.L. Pentose phosphates in nucleoside interconversion and catabolism. FEBS J. 2006, 273, 1089–1101. [Google Scholar] [CrossRef]
- Li, R.; Ke, M.; Qi, M.; Han, Z.; Cao, Y.; Deng, Z.; Qian, J.; Yang, Y.; Gu, C. G6PD promotes cell proliferation and dexamethasone resistance in multiple myeloma via increasing anti-oxidant production and activating Wnt/beta-catenin pathway. Exp. Hematol. Oncol. 2022, 11, 77. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nangia-Makker, P.; Ahrens, M.; Purandare, N.; Aras, S.; Li, J.; Gurdziel, K.; Jang, H.; Kim, S.; Shekhar, M.P. Inter-Relationship Between Melanoma Vemurafenib Tolerance Thresholds and Metabolic Pathway Choice. Cells 2025, 14, 923. https://doi.org/10.3390/cells14120923
Nangia-Makker P, Ahrens M, Purandare N, Aras S, Li J, Gurdziel K, Jang H, Kim S, Shekhar MP. Inter-Relationship Between Melanoma Vemurafenib Tolerance Thresholds and Metabolic Pathway Choice. Cells. 2025; 14(12):923. https://doi.org/10.3390/cells14120923
Chicago/Turabian StyleNangia-Makker, Pratima, Madison Ahrens, Neeraja Purandare, Siddhesh Aras, Jing Li, Katherine Gurdziel, Hyejeong Jang, Seongho Kim, and Malathy P Shekhar. 2025. "Inter-Relationship Between Melanoma Vemurafenib Tolerance Thresholds and Metabolic Pathway Choice" Cells 14, no. 12: 923. https://doi.org/10.3390/cells14120923
APA StyleNangia-Makker, P., Ahrens, M., Purandare, N., Aras, S., Li, J., Gurdziel, K., Jang, H., Kim, S., & Shekhar, M. P. (2025). Inter-Relationship Between Melanoma Vemurafenib Tolerance Thresholds and Metabolic Pathway Choice. Cells, 14(12), 923. https://doi.org/10.3390/cells14120923