
Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Importance of Understanding Primary and Secondary Damage Mechanisms
2.1. Primary Damage Mechanisms
2.1.1. Microvascular Damage
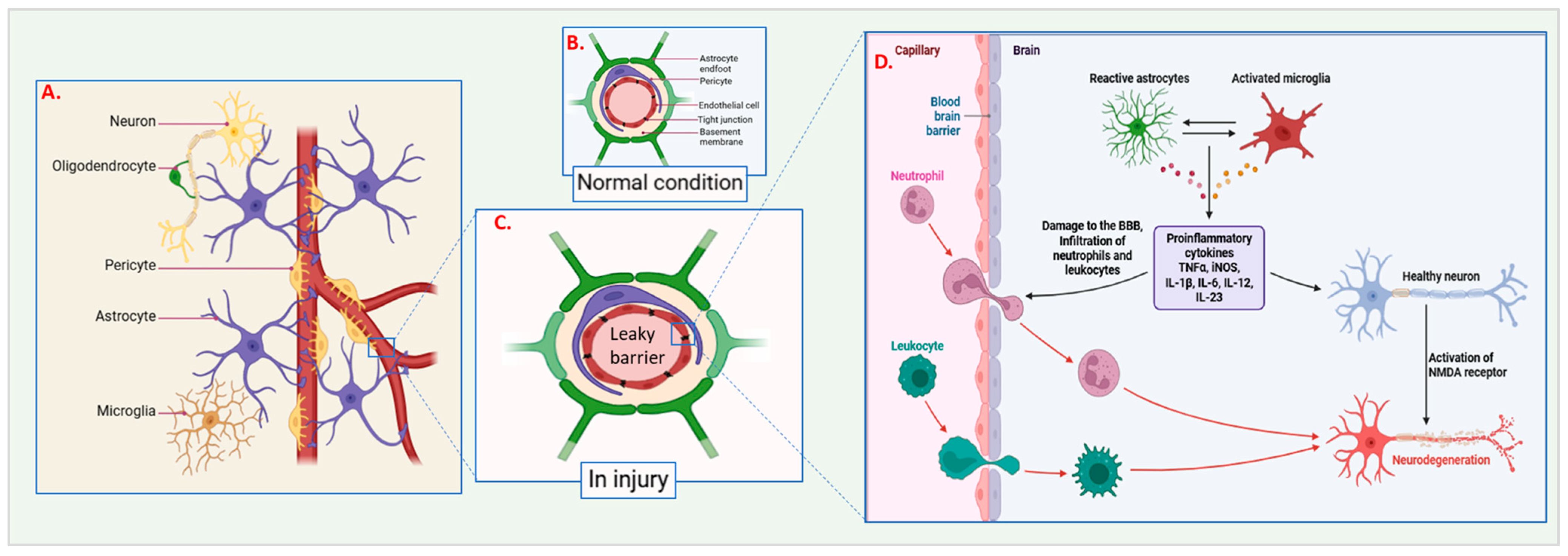
2.1.2. Mechanical Forces and Blood–Brain Barrier Disruption During Injury
2.1.3. Cell Membrane Damage
2.2. Secondary Damage Mechanisms
2.2.1. Ionic Imbalance and Calcium Overload
2.2.2. Mitochondrial Dysfunction and Stress Reactions
2.2.3. Excitability, Toxicity, and Chronic Inflammation
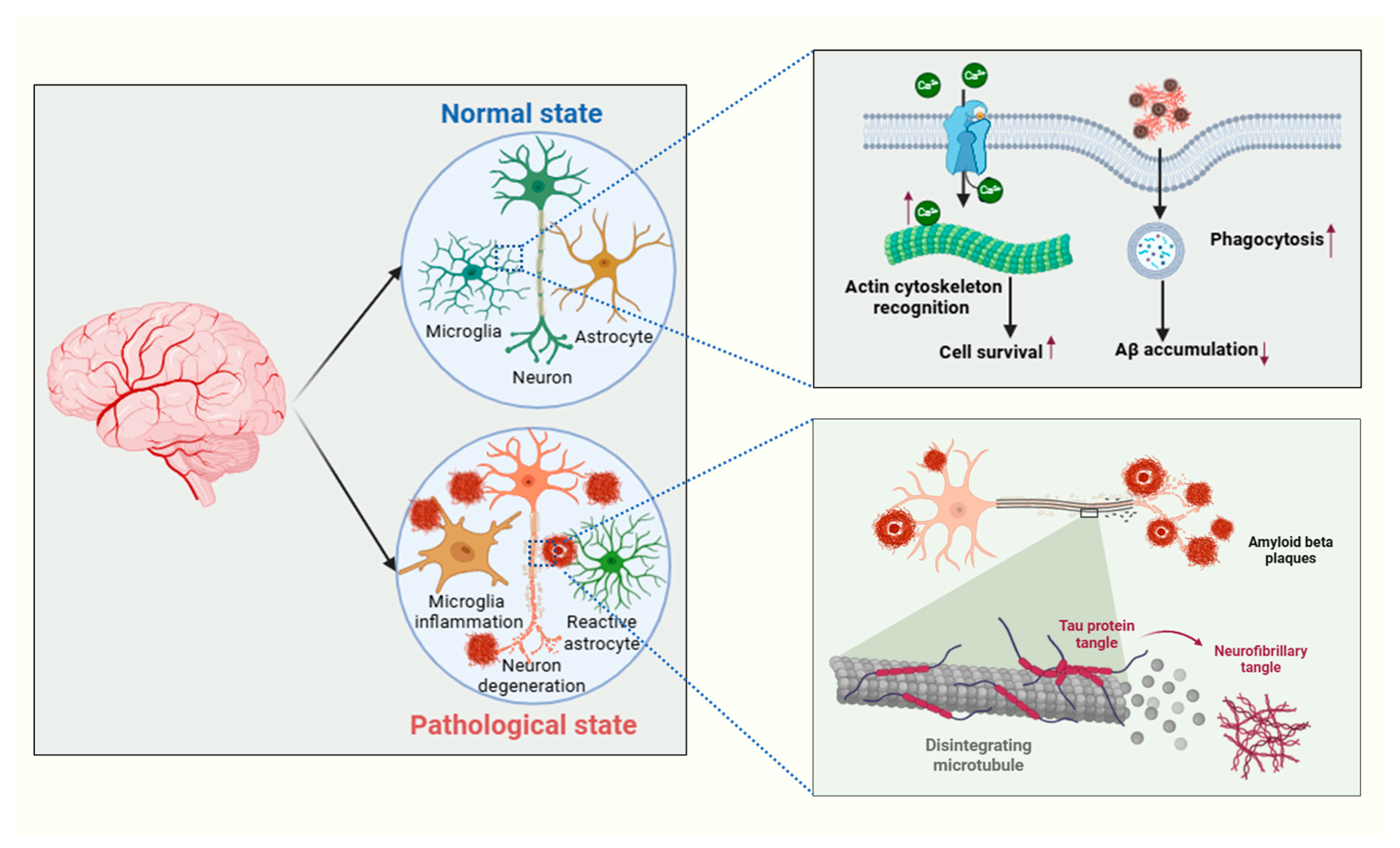
3. Neurodegenerative Pathways
3.1. Role of Tau Protein, Aβ Plaques, TDP-43, and α-Synuclein Deposits
3.2. Mechanisms Leading to Neurodegeneration
4. Inflammatory Response
4.1. Release of Cytokines and Chemokines
4.2. Activation of Astrocytes and Microglia
4.3. Recruitment of Circulating Immune Cells
5. CNS Repair Mechanisms
5.1. Formation of New Synapses
5.2. Neural Repair Processes
6. Role of Microglia in Injury and Repair
6.1. M1-like Microglia and Pro-Inflammatory Responses
6.2. M2-like Microglia and Anti-Inflammatory Responses
6.3. Phagocytic Activity and Tissue Repair
7. Therapeutic Implications
7.1. Targeting Inflammatory Pathways
7.2. Enhancing Repair Mechanisms
7.3. Potential for Neuroprotective Strategies
7.4. Disease-Tailored Therapeutic Approaches
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tam, R.Y.; Fuehrmann, T.; Mitrousis, N.; Shoichet, M.S. Regenerative Therapies for Central Nervous System Diseases: A Biomaterials Approach. Neuropsychopharmacology 2014, 39, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.G.; Hunyara, J.L.; Hamilton, N.R.; Kolodkin, A.L.; Huberman, A.D. Central nervous system regeneration. Cell 2022, 185, 77–94. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Lucke-Wold, B.P.; Turner, R.C.; Huber, J.D.; Rosen, C.L.; Simpkins, J.W. Role of Microvascular Disruption in Brain Damage from Traumatic Brain Injury. Compr. Physiol. 2015, 5, 1147–1160. [Google Scholar] [CrossRef]
- Price, L.; Wilson, C.; Grant, G. Blood-Brain Barrier Pathophysiology Following Traumatic Brain Injury; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Li, Y.; Song, J.; Liu, X.; Zhang, M.; An, J.; Sun, P.; Li, D.; Jin, T.; Wang, J. High expression of STIM1 in the early stages of diffuse axonal injury. Brain Res. 2013, 1495, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Huang, T.-H.; Lai, M.-C.; Chen, Y.-S.; Huang, C.-W. The Roles of Glutamate Receptors and Their Antagonists in Status Epilepticus, Refractory Status Epilepticus, and Super-Refractory Status Epilepticus. Biomedicines 2023, 11, 686. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Miller, M.A.; Zachary, J.F. Mechanisms and Morphology of Cellular Injury, Adaptation, and Death. In Pathologic Basis of Veterinary Disease; Elsevier: Amsterdam, The Netherlands, 2017; pp. 2–43.e19. [Google Scholar]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Khatri, N.; Sumadhura, B.; Kumar, S.; Kaundal, R.K.; Sharma, S.; Datusalia, A.K. The Complexity of Secondary Cascade Consequent to Traumatic Brain Injury: Pathobiology and Potential Treatments. Curr. Neuropharmacol. 2021, 19, 1984–2011. [Google Scholar] [CrossRef] [PubMed]
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.K. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm. Sin. B 2025, 15, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Wan, B.; Gong, G.; Yin, J. ROS: Executioner of regulating cell death in spinal cord injury. Front. Immunol. 2024, 15, 1330678. [Google Scholar] [CrossRef]
- Abbott, N.J. Dynamics of CNS Barriers: Evolution, Differentiation, and Modulation. Cell Mol. Neurobiol. 2005, 25, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Tepper, D.; Leonard, A. Matrix Metalloproteinases and Blood-Brain Barrier Disruption in Acute Ischemic Stroke. Front. Neurol. 2013, 4, 32. [Google Scholar] [CrossRef]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef]
- Vaughn, L.; Beckel, N.; Walters, P. Severe burn injury, burn shock, and smoke inhalation injury in small animals. Part 2: Diagnosis, therapy, complications, and prognosis. J. Vet. Emerg. Crit. Care 2012, 22, 187–200. [Google Scholar] [CrossRef]
- Song, X.-M.; Li, J.-G.; Wang, Y.-L.; Liang, H.; Huang, Y.; Yuan, X.; Zhou, Q.; Zhang, Z.Z. Effect of vagus nerve stimulation on thermal injury in rats. Burns 2010, 36, 75–81. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Z.; Yang, G.; Yang, D.; Ding, J.; Chen, H.; Yuan, F.; Tian, H.L. Sesamin alleviates blood-brain barrier disruption in mice with experimental traumatic brain injury. Acta Pharmacol. Sin. 2017, 38, 1445–1455. [Google Scholar] [CrossRef]
- Salimi, H.; Klein, R.S. Disruption of the Blood-Brain Barrier During Neuroinflammatory and Neuroinfectious Diseases. In Neuroimmune Diseases; Springer: Berlin/Heidelberg, Germany, 2019; pp. 195–234. [Google Scholar]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhang, C.; Chen, A.; Wang, H.; Zhou, Y.; Li, Y.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Armaly, Z.; Heyman, S.N. Glycocalyx Degradation in Ischemia-Reperfusion Injury. Am. J. Pathol. 2020, 190, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Jullienne, A.; Obenaus, A.; Ichkova, A.; Savona-Baron, C.; Pearce, W.J.; Badaut, J. Chronic cerebrovascular dysfunction after traumatic brain injury. J. Neurosci. Res. 2016, 94, 609–622. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef]
- Benarroch, E. What Are the Roles of Pericytes in the Neurovascular Unit and Its Disorders? Neurology 2023, 100, 970–977. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef]
- Patel, J.C.; Singh, A.; Tulswani, R.; Sharma, Y.K.; Khurana, P.; Ragumani, S. Identification of VEGFA-centric temporal hypoxia-responsive dynamic cardiopulmonary network biomarkers. Life Sci. 2021, 281, 119718. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The Pericyte: A Forgotten Cell Type with Important Implications for <scp>A</scp> lzheimer’s Disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in human central nervous system diseases: A frontier for new therapies. Signal Transduct. Target. Ther. 2023, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; de Jesus Perez, V.A. Hiding in Plain Sight: The Basement Membrane in Pulmonary Vascular Remodeling. Am. J. Respir. Cell Mol. Biol. 2020, 63, 13–14. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Coppini, A.; Falconieri, A.; Mualem, O.; Nasrin, S.R.; Roudon, M.; Saper, G.; Hess, H.; Kakugo, A.; Raffa, V.; Shefi, O. Can repetitive mechanical motion cause structural damage to axons? Front. Mol. Neurosci. 2024, 17, 1371738. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus. 2007, 22, E2. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.; Harris, A.; Wudunn, D.; Kheradiya, N.; Siesky, B. Dysfunctional regulation of ocular blood flow: A risk factor for glaucoma? Clin. Ophthalmol. 2008, 2, 849–861. [Google Scholar] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Kulbacka, J.; Choromańska, A.; Rossowska, J.; Weżgowiec, J.; Saczko, J.; Rols, M.-P. Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. Adv. Anat. Embryol. Cell Biol. 2017, 227, 39–58. [Google Scholar]
- Pirahanchi, Y.; Jessu, R.; Aeddula, N.R. Physiology, Sodium Potassium Pump. In Stat Pearls; Stat Pearls: Petersburg, FL, USA, 2025. [Google Scholar]
- Petkov, G.V. Ion Channels. In Pharmacology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 387–427. [Google Scholar]
- Olney, J.W.; Price, M.T.; Samson, L.; Labruyere, J. The role of specific ions in glutamate neurotoxicity. Neurosci. Lett. 1986, 65, 65–71. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Kristiaán, T.; Siesjö, B.K. Calcium in Ischemic Cell Death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W. Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic-cell damage. Med. Hypotheses 1996, 47, 283–288. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and Mitochondrial Reactive Oxygen Species Generation: How to Read the Facts. J. Alzheimer’s Dis. 2010, 20, S413–S426. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Molina, A.; Bretheau, F.; Boisvert, A.; Bélanger, D.; Lacroix, S. Constitutive DAMPs in CNS injury: From preclinical insights to clinical perspectives. Brain Behav. Immun. 2024, 122, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Sathe, K.; Maetzler, W.; Lang, J.D.; Mounsey, R.B.; Fleckenstein, C.; Martin, H.L.; Schulte, C.; Mustafa, S.; Synofzik, M.; Vukovic, Z.; et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-α pathway. Brain 2012, 135, 3336–3347. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.M.; González-Gallego, J. Manganese Superoxide Dismutase and Oxidative Stress Modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar] [PubMed]
- Milatovic, D.; Zaja-Milatovic, S.; Gupta, R.C. Biomarkers of oxidative/nitrosative stress and neurotoxicity. In Biomarkers in Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 863–881. [Google Scholar]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Shukla, M.; Mani, K.V.; Deepshikha Shukla, S.; Kapoor, N. Moderate noise associated oxidative stress with concomitant memory impairment, neuro-inflammation and neurodegeneration. Brain Behav. Immun. Health 2020, 5, 100089. [Google Scholar] [CrossRef]
- Zong, B.; Yu, F.; Zhang, X.; Pang, Y.; Zhao, W.; Sun, P.; Li, L. Mechanosensitive Piezo1 channel in physiology and pathophysiology of the central nervous system. Ageing Res. Rev. 2023, 90, 102026. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, K.; Laskowitz, D.T. Cellular and Molecular Mechanisms of Secondary Neuronal Injury following Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Song, M.; Yu, S.P. Ionic Regulation of Cell Volume Changes and Cell Death after Ischemic Stroke. Transl. Stroke Res. 2014, 5, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.; Waxman, S.; Ransom, B. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na(+)-Ca2+ exchanger. J. Neurosci. 1992, 12, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.D.; Farkas, E.; Hartings, J.A.; Brennan, K.C.; Herreras, O.; Müller, M.; Kirov, S.A.; Ayata, C.; Ollen-Bittle, N.; Reiffurth, C.; et al. Questioning Glutamate Excitotoxicity in Acute Brain Damage: The Importance of Spreading Depolarization. Neurocrit. Care 2022, 37, 11–30. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Nicosia, N.; Giovenzana, M.; Misztak, P.; Mingardi, J.; Musazzi, L. Glutamate-Mediated Excitotoxicity in the Pathogenesis and Treatment of Neurodevelopmental and Adult Mental Disorders. Int. J. Mol. Sci. 2024, 25, 6521. [Google Scholar] [CrossRef]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef]
- Berliocchi, L.; Bano, D.; Nicotera, P. Ca2+ signals and death programmes in neurons. Philos. Trans. R. Soc. B: Biol. Sci. 2005, 360, 2255–2258. [Google Scholar] [CrossRef]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef]
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-Mediated Signaling Mechanisms in Neuronal Injury and Neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimers Dis. Parkinsonism 2017, 7, 374. [Google Scholar] [CrossRef]
- De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. [Google Scholar] [CrossRef]
- Cheng, G.; Kong, R.; Zhang, L.; Zhang, J. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Nguyen, A.; Patel, A.B.; Kioutchoukova, I.P.; Diaz, M.J.; Lucke-Wold, B. Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics. Oxygen 2023, 3, 163–178. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.; Redondo-Flórez, L.; Beltrán-Velasco, A.; Ramos-Campo, D.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Yang, L.; Youngblood, H.; Wu, C.; Zhang, Q. Mitochondria as a target for neuroprotection: Role of methylene blue and photobiomodulation. Transl. Neurodegener. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Caponio, D.; Veverová, K.; Zhang, S.; Shi, L.; Wong, G.; Vyhnalek, M.; Fang, E.F. Compromised autophagy and mitophagy in brain ageing and Alzheimer’s diseases. Aging Brain 2022, 2, 100056. [Google Scholar] [CrossRef] [PubMed]
- Khot, M.; Sood, A.; Tryphena, K.P.; Khan, S.; Srivastava, S.; Singh, S.B.; Khatri, D.K. NLRP3 inflammasomes: A potential target to improve mitochondrial biogenesis in Parkinson’s disease. Eur. J. Pharmacol. 2022, 934, 175300. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Thapak, P.; Gomez-Pinilla, F. The bioenergetics of traumatic brain injury and its long-term impact for brain plasticity and function. Pharmacol. Res. 2024, 208, 107389. [Google Scholar] [CrossRef] [PubMed]
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflamm. 2018, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal Damage in Brain Inflammation. Arch. Neurol. 2007, 64, 185. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.L.; Stone, I.M.; Goss, A.; Rau, T.; Rova, C.; Poulsen, D.J. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia–ischemia. Neuroscience 2008, 155, 1204–1211. [Google Scholar] [CrossRef]
- Yang, J.; Vitery Mdel, C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef]
- Arranz, A.M.; Gottlieb, M.; Pérez-Cerdá, F.; Matute, C. Increased expression of glutamate transporters in subcortical white matter after transient focal cerebral ischemia. Neurobiol. Dis. 2010, 37, 156–165. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Shukla, M.; Patel, J.C.; Mani, K.V.; Nayak, D.; Shukla, M.; Sinha, D.; Chakraborty, S.; Kapoor, N. Impact of aircraft noise on hearing and physiological health using cognitive and physiological evaluations in military personnel. Discov. Med. 2025, 2, 63. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Theophanous, S.; Sargiannidou, I.; Kleopa, K.A. Glial Cells as Key Regulators in Neuroinflammatory Mechanisms Associated with Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 9588. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, A.; Albalkhi, I.; Magableh, H.M.; Saleh, T.; Alkattan, K.; Yaqinuddin, A. Tackling the glial scar in spinal cord regeneration: New discoveries and future directions. Front. Cell Neurosci. 2023, 17, 1180825. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191, Erratum in Nat. Rev. Neurol. 2017, 13, 572. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; You, H.; Hu, X.; Luo, Y.; Zhang, Z.; Song, Y.; An, J.; Lu, H. Microglia–Astrocyte Interaction in Neural Development and Neural Pathogenesis. Cells 2023, 12, 1942. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid Beta in Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12924. [Google Scholar] [CrossRef]
- Zeng, J.; Luo, C.; Jiang, Y.; Hu, T.; Lin, B.; Xie, Y.; Lan, J.; Miao, J. Decoding TDP-43: The molecular chameleon of neurodegenerative diseases. Acta Neuropathol. Commun. 2024, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: From overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Lee, H.-J.; Choi, C.; Lee, S.-J. Membrane-bound α-Synuclein Has a High Aggregation Propensity and the Ability to Seed the Aggregation of the Cytosolic Form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Wen, J.-H.; He, X.-H.; Feng, Z.-S.; Li, D.-Y.; Tang, J.-X.; Liu, H.-F. Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination. Int. J. Mol. Sci. 2023, 24, 8593. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A Crucial Factor in Alzheimer’s Disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Klemmensen, M.M.; Borrowman, S.H.; Pearce, C.; Pyles, B.; Chandra, B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 2024, 21, e00292. [Google Scholar] [CrossRef] [PubMed]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Houldsworth, A. Role of oxidative stress in neurodegenerative disorders: A review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2023, 6, fcad356. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Shukla, M.; Roy, K.; Kaur, C.; Nayak, D.; Mani, K.V.; Shukla, S.; Kapoor, N. Attenuation of adverse effects of noise induced hearing loss on adult neurogenesis and memory in rats by intervention with Adenosine A2A receptor agonist. Brain Res. Bull. 2019, 147, 47–57. [Google Scholar] [CrossRef]
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 Inclusion Bodies Formed in Bacteria Are Structurally Amorphous, Non-Amyloid and Inherently Toxic to Neuroblastoma Cells. PLoS ONE 2014, 9, e86720. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 1–20. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 1–20. [Google Scholar] [CrossRef]
- Aggrey, A.A.; Srivastava, K.; Ture, S.; Field, D.J.; Morrell, C.N. Platelet Induction of the Acute-Phase Response Is Protective in Murine Experimental Cerebral Malaria. J. Immunol. 2013, 190, 4685–4691. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive Gliosis and the Multicellular Response to CNS Damage and Disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimer’s Dement. 2022, 18, 2327–2340. [Google Scholar] [CrossRef]
- Kennedy, R.H.; Silver, R. Neuroimmune Signaling: Cytokines and the Central Nervous System. In Neuroscience in the 21st Century; Springer: New York, NY, USA, 2016; pp. 883–922. [Google Scholar]
- Zipp, F.; Bittner, S.; Schafer, D.P. Cytokines as emerging regulators of central nervous system synapses. Immunity 2023, 56, 914–925. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Fetsko, A.R.; Sebo, D.J.; Budzynski, L.B.; Scharbarth, A.; Taylor, M.R. IL-1β disrupts blood-brain barrier development by inhibiting endothelial Wnt/β-catenin signaling. bioRxiv 2023, 12, 569943. [Google Scholar] [CrossRef]
- Yang, J.; Ran, M.; Li, H.; Lin, Y.; Ma, K.; Yang, Y.; Fu, X.; Yang, S. New insight into neurological degeneration: Inflammatory cytokines and blood–brain barrier. Front. Mol. Neurosci. 2022, 15, 1013933. [Google Scholar] [CrossRef]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Caldito, N. Role of tumor necrosis factor-alpha in the central nervous system: A focus on autoimmune disorders. Front. Immunol. 2023, 14, 1213448. [Google Scholar] [CrossRef] [PubMed]
- Kummer, K.K.; Zeidler, M.; Kalpachidou, T.; Kress, M. Role of IL-6 in the regulation of neuronal development, survival and function. Cytokine 2021, 144, 155582. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef]
- Xin, Y.; Tian, M.; Deng, S.; Li, J.; Yang, M.; Gao, J.; Pei, X.; Wang, Y.; Tan, J.; Zhao, F.; et al. The Key Drivers of Brain Injury by Systemic Inflammatory Responses after Sepsis: Microglia and Neuroinflammation. Mol. Neurobiol. 2023, 60, 1369–1390. [Google Scholar] [CrossRef]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of Chemokines in CNS Health and Pathology: A Focus on the CCL2/CCR2 and CXCL8/CXCR2 Networks. J. Cereb. Blood Flow. Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Franke, H.; Illes, P. Nucleotide signaling in astrogliosis. Neurosci. Lett. 2014, 565, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Zhang, Y.; Ning, B. Reactive Astrocytes in Central Nervous System Injury: Subgroup and Potential Therapy. Front. Cell Neurosci. 2021, 15, 792764. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K.W. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Cao, Y.; Yang, J. Astrocyte-Mediated Neuroinflammation in Neurological Conditions. Biomolecules 2024, 14, 1204. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S.; Müller, V. From Homeostasis to Neuroinflammation: Insights into Cellular and Molecular Interactions and Network Dynamics. Cells 2025, 14, 54. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.; Chen, G.; Cui, S. Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front. Cell Neurosci. 2020, 14, 78. [Google Scholar]
- Anthony, D.C.; Couch, Y. The systemic response to CNS injury. Exp. Neurol. 2014, 258, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Zhu, C. Rolling Cell Adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 118, 6743–6751. [Google Scholar] [CrossRef]
- Gao, Y.-J.; Ji, R.-R. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef]
- Wang, J.; Bian, L.; Du, Y.; Wang, D.; Jiang, R.; Lu, J.; Zhao, X. The roles of chemokines following intracerebral hemorrhage in animal models and humans. Front. Mol. Neurosci. 2023, 15, 1091498. [Google Scholar] [CrossRef]
- Zhou, C.; Gao, Y.; Ding, P.; Wu, T.; Ji, G. The role of CXCL family members in different diseases. Cell Death Discov. 2023, 9, 212. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Nagappan, P.G.; Chen, H.; Wang, D.-Y. Neuroregeneration and plasticity: A review of the physiological mechanisms for achieving functional recovery postinjury. Mil. Med. Res. 2020, 7, 30. [Google Scholar] [CrossRef]
- Holt, R.L.; Mikati, M.A. Care for Child Development: Basic Science Rationale and Effects of Interventions. Pediatr. Neurol. 2011, 44, 239–253. [Google Scholar] [CrossRef]
- Marzola, P.; Melzer, T.; Pavesi, E.; Gil-Mohapel, J.; Brocardo, P.S. Exploring the Role of Neuroplasticity in Development, Aging, and Neurodegeneration. Brain Sci. 2023, 13, 1610. [Google Scholar] [CrossRef] [PubMed]
- Bitar, L.; Puig, B.; Oertner, T.G.; Dénes, Á.; Magnus, T. Changes in Neuroimmunological Synapses During Cerebral Ischemia. Transl Stroke Res. 2024. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 6, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hua, F.; Yang, D.; Lin, Y.; Zhang, L.; Ying, J.; Sheng, H.; Wang, X. Roles of neuroligins in central nervous system development: Focus on glial neuroligins and neuron neuroligins. J. Transl. Med. 2022, 20, 418. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Roles of glial cells in synapse development. Cell. Mol. Life Sci. 2009, 66, 2037–2047. [Google Scholar] [CrossRef]
- Araki, T.; Ikegaya, Y.; Koyama, R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur. J. Neurosci. 2021, 54, 5880–5901. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ying, J.; Wang, X.; Zheng, Q.; Zhao, T.; Yoon, S.; Yu, W.; Yang, D.; Fang, Y.; Hua, F. Astrocytes in Neural Circuits: Key Factors in Synaptic Regulation and Potential Targets for Neurodevelopmental Disorders. Front. Mol. Neurosci. 2021, 14, 729273. [Google Scholar] [CrossRef]
- Cortes, D.; Pera, M.F. The genetic basis of inter-individual variation in recovery from traumatic brain injury. NPJ Regen. Med. 2021, 6, 5. [Google Scholar] [CrossRef]
- Xu, X.; Warrington, A.E.; Bieber, A.J.; Rodriguez, M. Enhancing CNS Repair in Neurological Disease. CNS Drugs 2011, 25, 555–573. [Google Scholar] [CrossRef]
- Mokarram, N.; Bellamkonda, R.V. Overcoming Endogenous Constraints on Neuronal Regeneration. IEEE Trans. Biomed. Eng. 2011, 58, 1900–1906. [Google Scholar] [CrossRef]
- van Niekerk, E.A.; Tuszynski, M.H.; Lu, P.; Dulin, J.N. Molecular and Cellular Mechanisms of Axonal Regeneration After Spinal Cord Injury. Mol. Cell. Proteom. 2016, 15, 394–408. [Google Scholar] [CrossRef]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Akram, R.; Anwar, H.; Javed, M.S.; Rasul, A.; Imran, A.; Malik, S.A.; Raza, C.; Khan, I.U.; Sajid, F.; Iman, T.; et al. Axonal Regeneration: Underlying Molecular Mechanisms and Potential Therapeutic Targets. Biomedicines 2022, 10, 3186. [Google Scholar] [CrossRef]
- Dickendesher, T.L.; Baldwin, K.T.; Mironova, Y.A.; Koriyama, Y.; Raiker, S.J.; Askew, K.L.; Wood, A.; Geoffroy, C.G.; Zheng, B.; Liepmann, C.D.; et al. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat. Neurosci. 2012, 15, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Selzer, M.E.; Li, S. Scar-mediated inhibition and CSPG receptors in the CNS. Exp. Neurol. 2012, 237, 370–378. [Google Scholar] [CrossRef]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 2018, 7, 4. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Burnside, E.R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 2019, 10, 3879. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K. Glial Cells Shape Pathology and Repair After Spinal Cord Injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Simons, M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron 2022, 110, 3549–3565. [Google Scholar] [CrossRef]
- Lucas, S.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [PubMed]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [PubMed]
- Keough, M.B.; Yong, V.W. Remyelination Therapy for Multiple Sclerosis. Neurotherapeutics 2013, 10, 44–54. [Google Scholar] [CrossRef]
- Tepavčević, V.; Lubetzki, C. Oligodendrocyte progenitor cell recruitment and remyelination in multiple sclerosis: The more, the merrier? Brain 2022, 145, 4178–4192. [Google Scholar] [CrossRef]
- Zveik, O.; Rechtman, A.; Ganz, T.; Vaknin-Dembinsky, A. The interplay of inflammation and remyelination: Rethinking MS treatment with a focus on oligodendrocyte progenitor cells. Mol. Neurodegener. 2024, 19, 53. [Google Scholar] [CrossRef]
- Yun, M. Changes in Regenerative Capacity through Lifespan. Int. J. Mol. Sci. 2015, 16, 25392–25432. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gianmarco, L.; Kukley, M. Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury. Cells 2023, 12, 1842. [Google Scholar] [CrossRef]
- Tosolini, A.P.; Mentis, G.Z.; Martin, J.H. Editorial: Dysfunction and Repair of Neural Circuits for Motor Control. Front. Mol. Neurosci. 2021, 14, 669824. [Google Scholar] [CrossRef]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair After Brain Injury: A Helping Hand From Biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef]
- Nordvall, G.; Forsell, P.; Sandin, J. Neurotrophin-targeted therapeutics: A gateway to cognition and more? Drug Discov. Today 2022, 27, 103318. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; O’Connor, M.; Wang, G.; Han, F. Brain-Derived Neurotrophic Factor and Its Potential Therapeutic Role in Stroke Comorbidities. Neural Plast. 2020, 2020, 1969482. [Google Scholar] [CrossRef]
- Rahimi Darehbagh, R.; Seyedoshohadaei, S.A.; Ramezani, R.; Rezaei, N. Stem cell therapies for neurological disorders: Current progress, challenges, and future perspectives. Eur. J. Med. Res. 2024, 29, 386. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.J.; Breakefield, X.O.; Sena-Esteves, M. Genetic therapy for the nervous system. Hum. Mol. Genet. 2011, 20, R28–R41. [Google Scholar] [CrossRef]
- Pena, S.A.; Iyengar, R.; Eshraghi, R.S.; Bencie, N.; Mittal, J.; Aljohani, A.; Mittal, R.; Eshraghi, A.A. Gene therapy for neurological disorders: Challenges and recent advancements. J. Drug Target. 2020, 28, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Patel, T.; Sugandh, F.; Dev, J.; Kumar, U.; Adeeb, M.; Kachhadia, M.P.; Puri, P.; Prachi, F.N.U.; Zaman, M.U. Innovative Approaches and Therapies to Enhance Neuroplasticity and Promote Recovery in Patients with Neurological Disorders: A Narrative Review. Cureus 2023, 15, e41914. [Google Scholar] [CrossRef]
- Guo, L.; Choi, S.; Bikkannavar, P.; Cordeiro, M.F. Microglia: Key Players in Retinal Ageing and Neurodegeneration. Front. Cell Neurosci. 2022, 16, 804782. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front. Neurol. 2023, 14, 1103416. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Fenn, A.M.; Henry, C.J.; Huang, Y.; Dugan, A.; Godbout, J.P. Lipopolysaccharide-induced interleukin (IL)-4 receptor-α expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav. Immun. 2012, 26, 766–777. [Google Scholar] [CrossRef]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, H.; Wen, J.; Ma, K.; Chen, D.; Chen, Z.; Huang, C. Regulation of microglial process elongation, a featured characteristic of microglial plasticity. Pharmacol. Res. 2019, 139, 286–297. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Mahmood, A.; Miron, V.E. Microglia as therapeutic targets for central nervous system remyelination. Curr. Opin. Pharmacol. 2022, 63, 102188. [Google Scholar] [CrossRef]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front. Cell Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.-Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef]
- Könnecke, H.; Bechmann, I. The Role of Microglia and Matrix Metalloproteinases Involvement in Neuroinflammation and Gliomas. Clin. Dev. Immunol. 2013, 2013, 914104. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K. Microglia mediated neuroinflammation in neurodegenerative diseases: A review on the cell signaling pathways involved in microglial activation. J. Neuroimmunol. 2023, 383, 578180. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Zhang, J. A richer and more diverse future for microglia phenotypes. Heliyon 2023, 9, e14713. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front Immunol 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wang, Y.; Stetler, A.R.; Leak, R.K.; Hu, X.; Chen, J. Phagocytic microglia and macrophages in brain injury and repair. CNS Neurosci. Ther. 2022, 28, 1279–1293. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Herzog, C.; Pons Garcia, L.; Keatinge, M.; Greenald, D.; Moritz, C.; Peri, F.; Herrgen, L. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Development 2019, 146, dev174698. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; D’Ambrosi, N. Microglial Pruning: Relevance for Synaptic Dysfunction in Multiple Sclerosis and Related Experimental Models. Cells 2021, 10, 686. [Google Scholar] [CrossRef] [PubMed]
- Mordelt, A.; de Witte, L.D. Microglia-mediated synaptic pruning as a key deficit in neurodevelopmental disorders: Hype or hope? Curr. Opin. Neurobiol. 2023, 79, 102674. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Zhai, X.; Xie, L.; Guo, Y.; Chen, C.; Li, Y.; Wang, F.; Zhu, Z.; Zheng, L.; et al. Microglial phagocytosis and regulatory mechanisms after stroke. J. Cereb. Blood Flow. Metab. 2022, 42, 1579–1596. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef]
- Millán Solano, M.V.; Salinas Lara, C.; Sánchez-Garibay, C.; Soto-Rojas, L.O.; Escobedo-Ávila, I.; Tena-Suck, M.L.; Ortíz-Butrón, R.; Choreño-Parra, J.A.; Romero-López, J.P.; Meléndez Camargo, M.E. Effect of Systemic Inflammation in the CNS: A Silent History of Neuronal Damage. Int. J. Mol. Sci. 2023, 24, 11902. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2008, 132, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Jakaria Md Kim, I.-S.; Kim, J.; Haque, M.d.E.; Choi, D.-K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef]
- Hanamsagar, R.; Hanke, M.L.; Kielian, T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012, 33, 333–342. [Google Scholar] [CrossRef]
- Soraci, L.; Gambuzza, M.E.; Biscetti, L.; Laganà, P.; Lo Russo, C.; Buda, A.; Barresi, G.; Corsonello, A.; Lattanzio, F.; Lorello, G.; et al. Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson’s disease: Mechanisms and therapeutic implications. J. Neurol. 2023, 270, 1346–1360. [Google Scholar] [CrossRef]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisäkk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Prakash, R.; Kumari, N.; Ali Khan, M.; Quaiyoom Khan, A.; Uddin, S.; Verma, S.; Robertson, A.A.; Boltze, J.; Raza, S.S.; et al. MCC950 reduces autophagy and improves cognitive function by inhibiting NLRP3-dependent neuroinflammation in a rat model of Alzheimer’s disease. Brain Behav. Immun. 2024, 116, 70–84. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Ashayeri Ahmadabad, R.; Mirzaasgari, Z.; Gorji, A.; Khaleghi Ghadiri, M. Toll-Like Receptor Signaling Pathways: Novel Therapeutic Targets for Cerebrovascular Disorders. Int. J. Mol. Sci. 2021, 22, 6153. [Google Scholar] [CrossRef]
- Rosa, J.M.; Farré-Alins, V.; Ortega, M.C.; Navarrete, M.; Lopez-Rodriguez, A.B.; Palomino-Antolín, A.; Fernández-López, E.; Vila-del Sol, V.; Decouty, C.; Narros-Fernández, P.; et al. TLR4 pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. Br. J. Pharmacol. 2021, 178, 3395–3413. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hua, X.; Kong, D.; Stein, D.; Hua, F. Role of Toll-like receptor mediated signaling in traumatic brain injury. Neuropharmacology 2019, 145, 259–267. [Google Scholar] [CrossRef]
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the NLRP3 Inflammasome Inhibitors in In Vivo Disease Models. Molecules 2021, 26, 4996. [Google Scholar] [CrossRef]
- Li, C.; Luo, Y.; Li, S. The roles of neural stem cells in myelin regeneration and repair therapy after spinal cord injury. Stem Cell Res. Ther. 2024, 15, 204. [Google Scholar] [CrossRef]
- Rust, R.; Nih, L.R.; Liberale, L.; Yin, H.; El Amki, M.; Ong, L.K.; Zlokovic, B.V. Brain repair mechanisms after cell therapy for stroke. Brain 2024, 147, 3286–3305. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, A.; Muramatsu, R. Molecular Mechanisms of Central Nervous System Axonal Regeneration and Remyelination: A Review. Int. J. Mol. Sci. 2020, 21, 8116. [Google Scholar] [CrossRef] [PubMed]
- Han, P.-P.; Han, Y.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Enriched environment-induced neuroplasticity in ischemic stroke and its underlying mechanisms. Front. Cell Neurosci. 2023, 17, 1210361. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yuan, M.; Guo, Y.-S.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. The role of enriched environment in neural development and repair. Front. Cell Neurosci. 2022, 16, 890666. [Google Scholar] [CrossRef]
- López-Muguruza, E.; Matute, C. Alterations of Oligodendrocyte and Myelin Energy Metabolism in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 12912. [Google Scholar] [CrossRef]
- Neumann, B.; Segel, M.; Chalut, K.J.; Franklin, R.J. Remyelination and ageing: Reversing the ravages of time. Mult. Scler. J. 2019, 25, 1835–1841. [Google Scholar] [CrossRef]
- De Gioia, R.; Biella, F.; Citterio, G.; Rizzo, F.; Abati, E.; Nizzardo, M.; Bresolin, N.; Comi, G.P.; Corti, S. Neural Stem Cell Transplantation for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3103. [Google Scholar] [CrossRef]
- Shao, A.; Tu, S.; Lu, J.; Zhang, J. Crosstalk between stem cell and spinal cord injury: Pathophysiology and treatment strategies. Stem Cell Res. Ther. 2019, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Regenerative Stem Cell Therapy for Neurodegenerative Diseases: An Overview. Int. J. Mol. Sci. 2021, 22, 2153. [Google Scholar] [CrossRef]
- Zayed, M.A.; Sultan, S.; Alsaab, H.O.; Yousof, S.M.; Alrefaei, G.I.; Alsubhi, N.H.; Alkarim , S.; Al Ghamdi, K.S.; Bagabir , S.A.; Jana , A.; et al. Stem-Cell-Based Therapy: The Celestial Weapon against Neurological Disorders. Cells 2022, 11, 3476. [Google Scholar] [CrossRef]
- Yang, L.; Liu, S.-C.; Liu, Y.-Y.; Zhu, F.-Q.; Xiong, M.-J.; Hu, D.-X.; Zhang, W.J. Therapeutic role of neural stem cells in neurological diseases. Front. Bioeng. Biotechnol. 2024, 12, 1329712. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.L.; Sajed, D.; Tuszynski, M.H. Axonal Regeneration through Regions of Chondroitin Sulfate Proteoglycan Deposition after Spinal Cord Injury: A Balance of Permissiveness and Inhibition. J. Neurosci. 2003, 23, 9276–9288. [Google Scholar] [CrossRef]
- Tan, C.L.; Andrews, M.R.; Kwok, J.C.F.; Heintz, T.G.P.; Gumy, L.F.; Fässler, R.; Fawcett, J.W. Kindlin-1 Enhances Axon Growth on Inhibitory Chondroitin Sulfate Proteoglycans and Promotes Sensory Axon Regeneration. J. Neurosci. 2012, 32, 7325–7335. [Google Scholar] [CrossRef]
- Zhai, J.; Kim, H.; Han, S.B.; Manire, M.; Yoo, R.; Pang, S.; Smith, G.M.; Son, Y.J. Co-targeting myelin inhibitors and CSPGs markedly enhances regeneration of GDNF-stimulated, but not conditioning-lesioned, sensory axons into the spinal cord. Elife 2021, 10, e63050. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, X.; Cao, T.; Kang, R.; Huang, L. Insights on therapeutic potential of clemastine in neurological disorders. Front. Mol. Neurosci. 2023, 16, 1279985. [Google Scholar] [CrossRef]
- Buccilli, B.; Alan, A.; Baha’, A.; Shahzad, A.; Almealawy, Y.; Chisvo, N.S.; Ennabe, M.; Weinand, M. Neuroprotection strategies in traumatic brain injury: Studying the effectiveness of different clinical approaches. Surg. Neurol. Int. 2024, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective Strategies for Neurological Disorders by Natural Products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Candelario-Jalil, E. Emerging neuroprotective strategies for the treatment of ischemic stroke: An overview of clinical and preclinical studies. Exp. Neurol. 2021, 335, 113518. [Google Scholar] [CrossRef]
- Yang, N.; Guan, Q.-W.; Chen, F.-H.; Xia, Q.-X.; Yin, X.-X.; Zhou, H.-H.; Mao, H.Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxid. Med. Cell Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef]
- Neves, D.; Salazar, I.L.; Almeida, R.D.; Silva, R.M. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023, 328, 121814. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic cell death regulation in neurons. FEBS J. 2019, 286, 3276–3298. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Ganie, M.R.; Khan, N.; Shukla, M.; Sood, S.; Devi, S.; Arora, P.; Kumar, M.; Najar, I.A.; Tang, J. Okanin alleviates symptoms of nociceptive-like responses in diabetic peripheral neuropathy in type 1 diabetic Wistar rats by regulating the AGEs/NF-κB/Nrf-2 pathway. J. Pharmacol. Sci. 2025, 157, 12–24. [Google Scholar] [CrossRef]
- Olivares, D.K.; Deshpande, V.; Shi, Y.K.; Lahiri, D.H.; Greig, N.T.; Rogers, J.; Huang, X. N-Methyl D-Aspartate (NMDA) Receptor Antagonists and Memantine Treatment for Alzheimer’s Disease, Vascular Dementia and Parkinson’s Disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Kamat, C.D.; Gadal, S.; Mhatre, M.; Williamson, K.S.; Pye, Q.N.; Hensley, K. Antioxidants in Central Nervous System Diseases: Preclinical Promise and Translational Challenges. J. Alzheimer’s Dis. 2008, 15, 473–493. [Google Scholar] [CrossRef]
- Nakajima, Y.; Inokuchi, Y.; Nishi, M.; Shimazawa, M.; Otsubo, K.; Hara, H. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008, 1226, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Dhani, S.; Zhao, Y.; Zhivotovsky, B. A long way to go: Caspase inhibitors in clinical use. Cell Death Dis. 2021, 12, 949. [Google Scholar] [CrossRef]
- Unnisa, A.; Greig, N.H.; Kamal, M.A. Inhibition of Caspase 3 and Caspase 9 Mediated Apoptosis: A Multimodal Therapeutic Target in Traumatic Brain Injury. Curr. Neuropharmacol. 2023, 21, 1001–1012. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar] [PubMed]
- Shastry, S.; Hu, J.; Ying, M.; Mao, X. Cell Therapy for Parkinson’s Disease. Pharmaceutics 2023, 15, 2656. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Hersh, C.M. Updates and advances in multiple sclerosis neurotherapeutics. Neurodegener. Dis. Manag. 2023, 13, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Berger, J.R.; Greenberg, B. The Evolution of Anti-CD20 Treatment for Multiple Sclerosis: Optimization of Antibody Characteristics and Function. CNS Drugs 2025, 39, 545–564. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, J.C.; Shukla, M.; Shukla, M. Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair. Cells 2025, 14, 918. https://doi.org/10.3390/cells14120918
Patel JC, Shukla M, Shukla M. Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair. Cells. 2025; 14(12):918. https://doi.org/10.3390/cells14120918
Chicago/Turabian StylePatel, Jai Chand, Meenakshi Shukla, and Manish Shukla. 2025. "Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair" Cells 14, no. 12: 918. https://doi.org/10.3390/cells14120918
APA StylePatel, J. C., Shukla, M., & Shukla, M. (2025). Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair. Cells, 14(12), 918. https://doi.org/10.3390/cells14120918