
Roles of Bile Acid-Activated Receptors in Monocytes-Macrophages and Dendritic Cells



Abstract

1. Introduction
2. Biological Synthesis, Transport, and Metabolism of BAs
3. BA-Activated Receptors
3.1. FXR
3.2. LXR
3.3. VDR
3.4. GPBAR1
3.5. Other BA-Activated Receptors
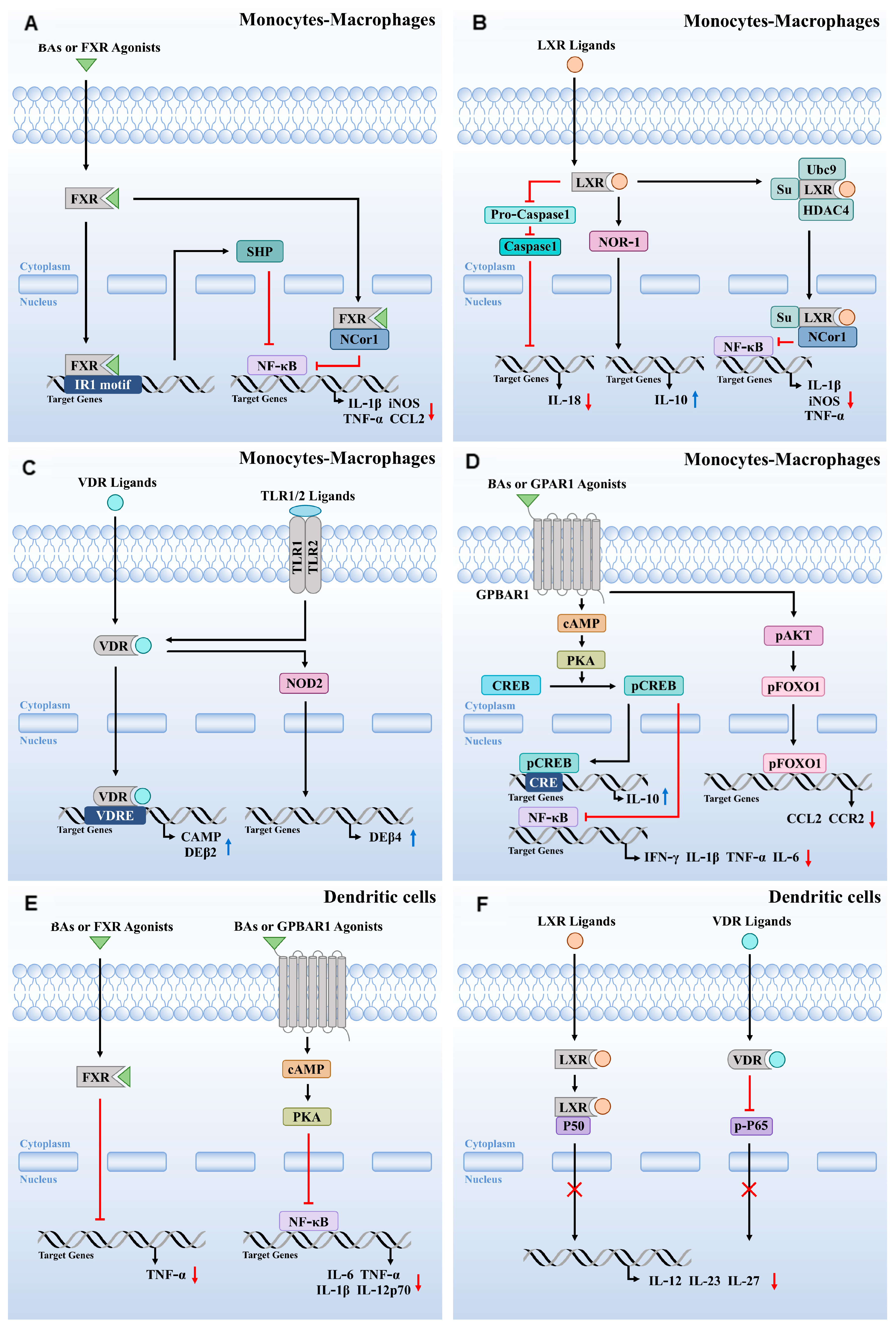
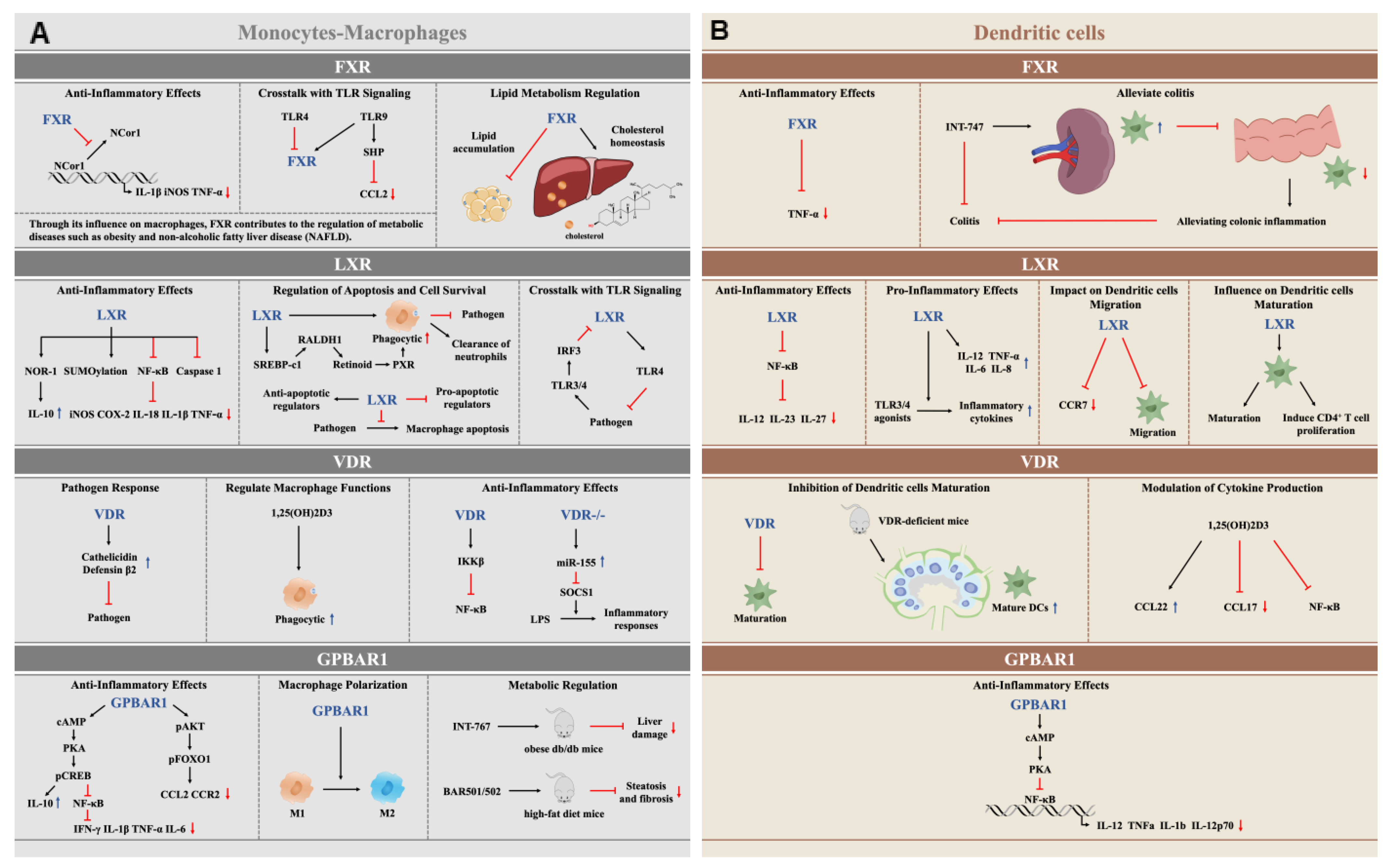
4. Roles of BA-Activated Receptors in Monocytes-Macrophages
4.1. The Anti-Inflammatory Function of FXR Activation in Monocytes-Macrophages
4.2. LXR Activation Regulates Inflammatory, Apoptotic, Phagocytic, and Pathogen Clearance Functions in Monocytes-Macrophages
4.3. VDR Activation Modulates Immune Responses in Monocytes-Macrophages
4.4. GPBAR1 Activation Modulates Immune Responses in Monocytes-Macrophages
5. Roles of BA-Activated Receptors in DCs
5.1. FXR and GPBAR1 Activation Modulates the Inflammation in DCs
5.2. LXR Activation Regulates Inflammation and the Immune Response in DCs
5.3. VDR Activation Regulates the Immune Response in DCs
6. Conclusions and Future Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, H.; Nie, R.; Shen, C. The role of bile acids in regulating glucose and lipid metabolism. Endocr. J. 2023, 70, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. Handb. Exp. Pharmacol. 2019, 256, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. Chenodeoxycholic Acid: An Update on Its Therapeutic Applications. Handb. Exp. Pharmacol. 2019, 256, 265–282. [Google Scholar] [CrossRef]
- Radreau, P.; Porcherot, M.; Ramiere, C.; Mouzannar, K.; Lotteau, V.; Andre, P. Reciprocal regulation of farnesoid X receptor alpha activity and hepatitis B virus replication in differentiated HepaRG cells and primary human hepatocytes. FASEB J. 2016, 30, 3146–3154. [Google Scholar] [CrossRef]
- Kong, F.; Saif, L.J.; Wang, Q. Roles of bile acids in enteric virus replication. Anim. Dis. 2021, 1, 2. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.L.; Stine, J.G.; Bisanz, J.E.; Okafor, C.D.; Patterson, A.D. Bile acids and the gut microbiota: Metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 2023, 21, 236–247. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, N.; Su, X.; Gao, Y.; Yang, R. Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis. Cells 2023, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Gui, W.; Koo, I.; Smith, P.B.; Allman, E.L.; Nichols, R.G.; Rimal, B.; Cai, J.; Liu, Q.; Patterson, A.D. The microbiome modulating activity of bile acids. Gut Microbes 2020, 11, 979–996. [Google Scholar] [CrossRef]
- Rimal, B.; Collins, S.L.; Tanes, C.E.; Rocha, E.R.; Granda, M.A.; Solanki, S.; Hoque, N.J.; Gentry, E.C.; Koo, I.; Reilly, E.R.; et al. Bile salt hydrolase catalyses formation of amine-conjugated bile acids. Nature 2024, 626, 859–863. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P.; Bhattacharyya, A.; Hunter, R.J.; Heidenreich, R.A.; Cherrington, N.J. Liver disease with altered bile acid transport in Niemann-Pick C mice on a high-fat, 1% cholesterol diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G300–G307. [Google Scholar] [CrossRef]
- Silvennoinen, R.; Quesada, H.; Kareinen, I.; Julve, J.; Kaipiainen, L.; Gylling, H.; Blanco-Vaca, F.; Escola-Gil, J.C.; Kovanen, P.T.; Lee-Rueckert, M. Chronic intermittent psychological stress promotes macrophage reverse cholesterol transport by impairing bile acid absorption in mice. Physiol. Rep. 2015, 3, e12402. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, H.; Li, Q. Hepatic macrophage niche: A bridge between HBV-mediated metabolic changes with intrahepatic inflammation. Front. Immunol. 2024, 15, 1414594. [Google Scholar] [CrossRef]
- Zheng, D.; Ge, K.; Qu, C.; Sun, T.; Wang, J.; Jia, W.; Zhao, A. Comparative profiling of serum, urine, and feces bile acids in humans, rats, and mice. Commun. Biol. 2024, 7, 641. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Fang, Y.; Hegazy, L.; Finck, B.N.; Elgendy, B. Recent Advances in the Medicinal Chemistry of Farnesoid X Receptor. J. Med. Chem. 2021, 64, 17545–17571. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Migliorati, M.; Cipriani, S.; Distrutti, E.; Santucci, L.; Fiorucci, S. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J. Immunol. 2009, 183, 6657–6666. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, H.M.; Voges, V.C.; Braun, L.C.; Sonnenberg, J.; Schwarz, D.; Korner, H.; Reinecke, H.; Sohrabi, Y. LXRalpha Regulates oxLDL-Induced Trained Immunity in Macrophages. Int. J. Mol. Sci. 2022, 23, 6166. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Song, C.; Hiipakka, R.A.; Liao, S. Selective activation of liver X receptor alpha by 6alpha-hydroxy bile acids and analogs. Steroids 2000, 65, 423–427. [Google Scholar] [CrossRef]
- Janousek, J.; Pilarova, V.; Macakova, K.; Nomura, A.; Veiga-Matos, J.; da Silva, D.D.; Remiao, F.; Saso, L.; Mala-Ladova, K.; Maly, J.; et al. Vitamin D: Sources, physiological role, biokinetics, deficiency, therapeutic use, toxicity, and overview of analytical methods for detection of vitamin D and its metabolites. Crit. Rev. Clin. Lab. Sci. 2022, 59, 517–554. [Google Scholar] [CrossRef]
- Lin, R. Crosstalk between Vitamin D Metabolism, VDR Signalling, and Innate Immunity. Biomed. Res. Int. 2016, 2016, 1375858. [Google Scholar] [CrossRef]
- Ishizawa, M.; Akagi, D.; Makishima, M. Lithocholic Acid Is a Vitamin D Receptor Ligand That Acts Preferentially in the Ileum. Int. J. Mol. Sci. 2018, 19, 1975. [Google Scholar] [CrossRef]
- Aggeletopoulou, I.; Thomopoulos, K.; Mouzaki, A.; Triantos, C. Vitamin D-VDR Novel Anti-Inflammatory Molecules-New Insights into Their Effects on Liver Diseases. Int. J. Mol. Sci. 2022, 23, 8465. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. Minireview: Vitamin D receptor: New assignments for an already busy receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Zheng, M.; Chen, Y.; Xiong, H. Update on the development of TGR5 agonists for human diseases. Eur. J. Med. Chem. 2024, 271, 116462. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Ikegami, T.; Honda, A. Reciprocal interactions between bile acids and gut microbiota in human liver diseases. Hepatol. Res. 2018, 48, 15–27. [Google Scholar] [CrossRef]
- Dutta, M.; Lim, J.J.; Cui, J.Y. Pregnane X Receptor and the Gut-Liver Axis: A Recent Update. Drug Metab. Dispos. 2022, 50, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Kurian, R.; Wang, H. Clinical Relevance of the Constitutive Androstane Receptor. Drug Metab. Dispos. 2022, 50, 1010–1018. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Ferrari, C.; Macchiarulo, A.; Costantino, G.; Pellicciari, R. Pharmacophore model for bile acids recognition by the FPR receptor. J. Comput. Aided Mol. Des. 2006, 20, 295–303. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Zhang, Y.; Liu, P.; Liu, M.; Zhang, M.; Wu, R. S1P/S1PR signaling pathway advancements in autoimmune diseases. Biomol. Biomed. 2023, 23, 922–935. [Google Scholar] [CrossRef]
- Yang, J.; Tang, X.; Liang, Z.; Chen, M.; Sun, L. Taurocholic acid promotes hepatic stellate cell activation via S1PR2/p38 MAPK/YAP signaling under cholestatic conditions. Clin. Mol. Hepatol. 2023, 29, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Carter, P.A.; Zhang, L.; Falcone, J.F.; Stengel, P.W.; Cohen, M.L.; Shannon, H.E.; Gomeza, J.; Wess, J.; Felder, C.C. Investigations into the physiological role of muscarinic M2 and M4 muscarinic and M4 receptor subtypes using receptor knockout mice. Life Sci. 2001, 68, 2473–2479. [Google Scholar] [CrossRef] [PubMed]
- Peretto, I.; Petrillo, P.; Imbimbo, B.P. Medicinal chemistry and therapeutic potential of muscarinic M3 antagonists. Med. Res. Rev. 2009, 29, 867–902. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, K.; Xiang, Y.; Yoshimura, T.; Su, S.; Zhu, J.; Bian, X.W.; Wang, J.M. New development in studies of formyl-peptide receptors: Critical roles in host defense. J. Leukoc. Biol. 2016, 99, 425–435. [Google Scholar] [CrossRef]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef]
- Sepe, V.; Distrutti, E.; Limongelli, V.; Fiorucci, S.; Zampella, A. Steroidal scaffolds as FXR and GPBAR1 ligands: From chemistry to therapeutical application. Future Med. Chem. 2015, 7, 1109–1135. [Google Scholar] [CrossRef]
- Adada, M.; Canals, D.; Hannun, Y.A.; Obeid, L.M. Sphingosine-1-phosphate receptor 2. FEBS J. 2013, 280, 6354–6366. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef]
- Yang, Z.; Koehler, A.N.; Wang, L. A Novel Small Molecule Activator of Nuclear Receptor SHP Inhibits HCC Cell Migration via Suppressing Ccl2. Mol. Cancer Ther. 2016, 15, 2294–2301. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Massafra, V.; Ijssennagger, N.; Plantinga, M.; Milona, A.; Ramos Pittol, J.M.; Boes, M.; van Mil, S.W. Splenic dendritic cell involvement in FXR-mediated amelioration of DSS colitis. Biochim. Biophys. Acta 2016, 1862, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Ishikiriyama, T.; Nakashima, H.; Endo-Umeda, K.; Nakashima, M.; Ito, S.; Kinoshita, M.; Ikarashi, M.; Makishima, M.; Seki, S. Contrasting functional responses of resident Kupffer cells and recruited liver macrophages to irradiation and liver X receptor stimulation. PLoS ONE 2021, 16, e0254886. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- He, K.; Dai, Z.Y.; Li, P.Z.; Zhu, X.W.; Gong, J.P. Association between liver X receptor-alpha and neuron-derived orphan nuclear receptor-1 in Kupffer cells of C57BL/6 mice during inflammation. Mol. Med. Rep. 2015, 12, 6098–6104. [Google Scholar] [CrossRef]
- Pourcet, B.; Gage, M.C.; Leon, T.E.; Waddington, K.E.; Pello, O.M.; Steffensen, K.R.; Castrillo, A.; Valledor, A.F.; Pineda-Torra, I. The nuclear receptor LXR modulates interleukin-18 levels in macrophages through multiple mechanisms. Sci. Rep. 2016, 6, 25481. [Google Scholar] [CrossRef]
- Endo-Umeda, K.; Nakashima, H.; Komine-Aizawa, S.; Umeda, N.; Seki, S.; Makishima, M. Liver X receptors regulate hepatic F4/80+CD11b+ Kupffer cells/macrophages and innate immune responses in mice. Sci. Rep. 2018, 8, 9281. [Google Scholar] [CrossRef]
- Fontaine, C.; Rigamonti, E.; Nohara, A.; Gervois, P.; Teissier, E.; Fruchart, J.C.; Staels, B.; Chinetti-Gbaguidi, G. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ. Res. 2007, 101, 40–49. [Google Scholar] [CrossRef]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Sohrabi, Y.; Sonntag, G.V.H.; Braun, L.C.; Lagache, S.M.M.; Liebmann, M.; Klotz, L.; Godfrey, R.; Kahles, F.; Waltenberger, J.; Findeisen, H.M. LXR Activation Induces a Proinflammatory Trained Innate Immunity-Phenotype in Human Monocytes. Front. Immunol. 2020, 11, 353. [Google Scholar] [CrossRef]
- Joseph, S.B.; Bradley, M.N.; Castrillo, A.; Bruhn, K.W.; Mak, P.A.; Pei, L.; Hogenesch, J.; O’Connell, R.M.; Cheng, G.; Saez, E.; et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 2004, 119, 299–309. [Google Scholar] [CrossRef]
- Bouttier, M.; Laperriere, D.; Memari, B.; Mangiapane, J.; Fiore, A.; Mitchell, E.; Verway, M.; Behr, M.A.; Sladek, R.; Barreiro, L.B.; et al. Alu repeats as transcriptional regulatory platforms in macrophage responses to M. tuberculosis infection. Nucleic Acids Res. 2016, 44, 10571–10587. [Google Scholar] [CrossRef]
- Matalonga, J.; Glaria, E.; Bresque, M.; Escande, C.; Carbo, J.M.; Kiefer, K.; Vicente, R.; Leon, T.E.; Beceiro, S.; Pascual-Garcia, M.; et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017, 18, 1241–1255. [Google Scholar] [CrossRef]
- Valledor, A.F.; Hsu, L.C.; Ogawa, S.; Sawka-Verhelle, D.; Karin, M.; Glass, C.K. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 17813–17818. [Google Scholar] [CrossRef] [PubMed]
- A-Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Morrell, E.D.; Gowdy, K.M.; McDonald, J.G.; Thompson, B.M.; Muse, G.; Martinez, J.; Thomas, S.; Mikacenic, C.; Nick, J.A.; et al. Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight 2020, 5, e137189. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Morrell, E.D.; McDonald, J.G.; Thompson, B.M.; Li, Y.; Birukov, K.G.; Birukova, A.A.; Stapleton, R.D.; Alejo, A.; Karmaus, P.W.; et al. 25-Hydroxycholesterol exacerbates vascular leak during acute lung injury. JCI Insight 2023, 8, e155448. [Google Scholar] [CrossRef]
- Rebe, C.; Raveneau, M.; Chevriaux, A.; Lakomy, D.; Sberna, A.L.; Costa, A.; Bessede, G.; Athias, A.; Steinmetz, E.; Lobaccaro, J.M.; et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res. 2009, 105, 393–401. [Google Scholar] [CrossRef]
- Sarang, Z.; Joos, G.; Garabuczi, E.; Ruhl, R.; Gregory, C.D.; Szondy, Z. Macrophages engulfing apoptotic cells produce nonclassical retinoids to enhance their phagocytic capacity. J. Immunol. 2014, 192, 5730–5738. [Google Scholar] [CrossRef]
- He, Q.; Ananaba, G.A.; Patrickson, J.; Pitts, S.; Yi, Y.; Yan, F.; Eko, F.O.; Lyn, D.; Black, C.M.; Igietseme, J.U.; et al. Chlamydial infection in vitamin D receptor knockout mice is more intense and prolonged than in wild-type mice. J. Steroid Biochem. Mol. Biol. 2013, 135, 7–14. [Google Scholar] [CrossRef]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Grad, R. Cod and the consumptive: A brief history of cod-liver oil in the treatment of pulmonary tuberculosis. Pharm. Hist. 2004, 46, 106–120. [Google Scholar]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Schenk, M.; Walker, V.P.; Dempsey, P.W.; Kanchanapoomi, M.; Wheelwright, M.; Vazirnia, A.; Zhang, X.; Steinmeyer, A.; Zugel, U.; et al. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS ONE 2009, 4, e5810. [Google Scholar] [CrossRef]
- Wang, T.T.; Dabbas, B.; Laperriere, D.; Bitton, A.J.; Soualhine, H.; Tavera-Mendoza, L.E.; Dionne, S.; Servant, M.J.; Bitton, A.; Seidman, E.G.; et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J. Biol. Chem. 2010, 285, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Umeda, N.; Endo-Umeda, K.; Nakashima, H.; Kato, S.; Seki, S.; Makishima, M. Frontline Science: Concanavalin A-induced acute hepatitis is attenuated in vitamin D receptor knockout mice with decreased immune cell function. J. Leukoc. Biol. 2019, 106, 791–801. [Google Scholar] [CrossRef]
- Shi, H.; Duan, J.; Wang, J.; Li, H.; Wu, Z.; Wang, S.; Wu, X.; Lu, M. 1,25(OH)(2)D(3) Promotes Macrophage Efferocytosis Partly by Upregulating ASAP2 Transcription via the VDR-Bound Enhancer Region and ASAP2 May Affect Antiviral Immunity. Nutrients 2022, 14, 4935. [Google Scholar] [CrossRef]
- Li, Y.C.; Chen, Y.; Liu, W.; Thadhani, R. MicroRNA-mediated mechanism of vitamin D regulation of innate immune response. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 81–86. [Google Scholar] [CrossRef]
- Meyer, V.; Saccone, D.S.; Tugizimana, F.; Asani, F.F.; Jeffery, T.J.; Bornman, L. Methylation of the Vitamin D Receptor (VDR) Gene, Together with Genetic Variation, Race, and Environment Influence the Signaling Efficacy of the Toll-like Receptor 2/1-VDR Pathway. Front. Immunol. 2017, 8, 1048. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Haselow, K.; Bode, J.G.; Wammers, M.; Ehlting, C.; Keitel, V.; Kleinebrecht, L.; Schupp, A.K.; Haussinger, D.; Graf, D. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J. Leukoc. Biol. 2013, 94, 1253–1264. [Google Scholar] [CrossRef]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchiano, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Fiorucci, C.; Marchiano, S.; Di Giorgio, C.; Bordoni, M.; Roselli, R.; Baldoni, M.; Distrutti, E.; Zampella, A.; et al. The Bile Acid Receptor GPBAR1 Modulates CCL2/CCR2 Signaling at the Liver Sinusoidal/Macrophage Interface and Reverses Acetaminophen-Induced Liver Toxicity. J. Immunol. 2020, 204, 2535–2551. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Neurath, M.F. Novel Insights into the Mechanisms of Gut Homing and Antiadhesion Therapies in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2017, 23, 617–627. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, S.; Shi, Y.; Wang, Q.; Wei, S.; Wang, P.; Cheng, F.; Auwerx, J.; Schoonjans, K.; Lu, L. TGR5/Cathepsin E signaling regulates macrophage innate immune activation in liver ischemia and reperfusion injury. Am. J. Transpl. 2021, 21, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, W.; Zhang, L.; Shi, C.; Zhou, J.; Wang, P.; Wang, H.; Shi, X.; Wei, S.; Wang, Q.; et al. TGR5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609060. [Google Scholar] [CrossRef]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef]
- Carino, A.; Cipriani, S.; Marchiano, S.; Biagioli, M.; Santorelli, C.; Donini, A.; Zampella, A.; Monti, M.C.; Fiorucci, S. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep. 2017, 7, 42801. [Google Scholar] [CrossRef] [PubMed]
- Hogenauer, K.; Arista, L.; Schmiedeberg, N.; Werner, G.; Jaksche, H.; Bouhelal, R.; Nguyen, D.G.; Bhat, B.G.; Raad, L.; Rauld, C.; et al. G-protein-coupled bile acid receptor 1 (GPBAR1, TGR5) agonists reduce the production of proinflammatory cytokines and stabilize the alternative macrophage phenotype. J. Med. Chem. 2014, 57, 10343–10354. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, C.; Huang, X.; Yi, S.; Pan, S.; Zhang, Y.; Yuan, G.; Cao, Q.; Ye, X.; Li, H. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021, 36, 109726. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef]
- Kiss, M.; Czimmerer, Z.; Nagy, L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J. Allergy Clin. Immunol. 2013, 132, 264–286. [Google Scholar] [CrossRef]
- Canavan, M.; McCarthy, C.; Larbi, N.B.; Dowling, J.K.; Collins, L.; O’Sullivan, F.; Hurley, G.; Murphy, C.; Quinlan, A.; Moloney, G.; et al. Activation of liver X receptor suppresses the production of the IL-12 family of cytokines by blocking nuclear translocation of NF-kappaBp50. Innate Immun. 2014, 20, 675–687. [Google Scholar] [CrossRef]
- Torocsik, D.; Barath, M.; Benko, S.; Szeles, L.; Dezso, B.; Poliska, S.; Hegyi, Z.; Homolya, L.; Szatmari, I.; Lanyi, A.; et al. Activation of liver X receptor sensitizes human dendritic cells to inflammatory stimuli. J. Immunol. 2010, 184, 5456–5465. [Google Scholar] [CrossRef]
- Ceroi, A.; Delettre, F.A.; Marotel, C.; Gauthier, T.; Asgarova, A.; Biichle, S.; Duperrier, A.; Mourey, G.; Perruche, S.; Lagrost, L.; et al. The anti-inflammatory effects of platelet-derived microparticles in human plasmacytoid dendritic cells involve liver X receptor activation. Haematologica 2016, 101, e72–e76. [Google Scholar] [CrossRef]
- Barragan, M.; Good, M.; Kolls, J.K. Regulation of Dendritic Cell Function by Vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef]
- Berer, A.; Stockl, J.; Majdic, O.; Wagner, T.; Kollars, M.; Lechner, K.; Geissler, K.; Oehler, L. 1,25-Dihydroxyvitamin D(3) inhibits dendritic cell differentiation and maturation in vitro. Exp. Hematol. 2000, 28, 575–583. [Google Scholar] [CrossRef]
- Griffin, M.D.; Lutz, W.; Phan, V.A.; Bachman, L.A.; McKean, D.J.; Kumar, R. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: A vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 6800–6805. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.; Amuchastegui, S.; Giarratana, N.; Daniel, K.C.; Vulcano, M.; Sozzani, S.; Adorini, L. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J. Immunol. 2007, 178, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, E.; Verlinden, L.; Giulietti, A.; Ramos-Lopez, E.; Branisteanu, D.D.; Ferreira, G.B.; Overbergh, L.; Verstuyf, A.; Bouillon, R.; Roep, B.O.; et al. The vitamin D receptor gene FokI polymorphism: Functional impact on the immune system. Eur. J. Immunol. 2007, 37, 395–405. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BA-Activated Receptors | Tissue Distributions | Cell Distributions | BA Ligands | Antagonists | References | |
|---|---|---|---|---|---|---|
| Nuclear receptors | FXR | liver, heart, kidneys, intestines | hepatocytes, cholangiocytes, liver sinusoidal cells, intestinal and liver endothelial cells, monocytes-macrophages, DCs, NK cells, NKT cells, T cells, B cells | CDCA > LCA > DCA > CA | UDCA, β-MCA, Gly-MCA | [17,18,46] |
| LXR | intestines, liver, kidneys, lungs, adipose tissue | hepatocytes, macrophages | 6α-hydroxylated BAs | - | [22,27] | |
| VDR | ileum, kidneys, liver, bones, skin, endocrine tissues | T cells, B cells, NK cells, DCs, monocytes-macrophages, non-parenchymal liver cells | DCA, LCA | - | [30,31] | |
| PXR | liver, intestines | hepatocytes | 3-keto-LCA, LCA, DCA | - | [36] | |
| CAR | liver, small intestine, gallbladder | hepatocytes | CA, LCA | - | [37] | |
| Cell membrane receptors | GPBAR1 (TGR5) | intestines, stomach, liver, spleen, gallbladder, adipose tissue | sinusoidal endothelial cells, cholangiocytes, hepatic stellate cells, monocytes-macrophages, DCs, NKs cells, NKT cells | TLCA > LCA > DCA > CDCA > CA | - | [33,46] |
| S1PR2 | Most tissues | hepatocytes | Taurine and glycine conjugated BAs | - | [38,47] | |
| M2 and M3 | heart, central nervous system | smooth muscle cells | DCA-LCA | - | [6] | |
| FPR1 | - | macrophages | - | DCA, CDCA | [39] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, H.; He, X.; Jiang, T.; Kong, F. Roles of Bile Acid-Activated Receptors in Monocytes-Macrophages and Dendritic Cells. Cells 2025, 14, 920. https://doi.org/10.3390/cells14120920
Jia H, He X, Jiang T, Kong F. Roles of Bile Acid-Activated Receptors in Monocytes-Macrophages and Dendritic Cells. Cells. 2025; 14(12):920. https://doi.org/10.3390/cells14120920
Chicago/Turabian StyleJia, Huilin, Xingli He, Tengfei Jiang, and Fanzhi Kong. 2025. "Roles of Bile Acid-Activated Receptors in Monocytes-Macrophages and Dendritic Cells" Cells 14, no. 12: 920. https://doi.org/10.3390/cells14120920
APA StyleJia, H., He, X., Jiang, T., & Kong, F. (2025). Roles of Bile Acid-Activated Receptors in Monocytes-Macrophages and Dendritic Cells. Cells, 14(12), 920. https://doi.org/10.3390/cells14120920